Microarray Comparative Genomic Hybridization Analysis of 59 Patients with Schizophrenia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CD8 T Cell Memory Transition and Maintenance of Functional Foxo1
FoxO1 Controls Effector-to-Memory Transition and Maintenance of Functional CD8 T Cell Memory This information is current as Melba Marie Tejera, Eui Ho Kim, Jeremy A. Sullivan, Erin of October 1, 2021. H. Plisch and M. Suresh J Immunol 2013; 191:187-199; Prepublished online 3 June 2013; doi: 10.4049/jimmunol.1300331 http://www.jimmunol.org/content/191/1/187 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2013/06/03/jimmunol.130033 Material 1.DC1 http://www.jimmunol.org/ References This article cites 52 articles, 7 of which you can access for free at: http://www.jimmunol.org/content/191/1/187.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on October 1, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology FoxO1 Controls Effector-to-Memory Transition and Maintenance of Functional CD8 T Cell Memory Melba Marie Tejera, Eui Ho Kim, Jeremy A. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

FIST/HIPK3: a Fas/FADD-Interacting Serine/Threonine Kinase That Induces FADD Phosphorylation and Inhibits Fas-Mediated Jun NH2-Terminal Kinase Activation
FIST/HIPK3: a Fas/FADD-interacting Serine/Threonine Kinase that Induces FADD Phosphorylation and Inhibits Fas-mediated Jun NH2-terminal Kinase Activation By Véronique Rochat-Steiner, Karin Becker, Olivier Micheau, Pascal Schneider, Kim Burns, and Jürg Tschopp From the Institute of Biochemistry, University of Lausanne, BIL Biomedical Research Center, CH- 1066 Epalinges, Switzerland Abstract Fas is a cell surface death receptor that signals apoptosis. Several proteins have been identified that bind to the cytoplasmic death domain of Fas. Fas-associated death domain (FADD), which couples Fas to procaspase-8, and Daxx, which couples Fas to the Jun NH2-terminal kinase pathway, bind independently to the Fas death domain. We have identified a 130-kD ki- nase designated Fas-interacting serine/threonine kinase/homeodomain-interacting protein ki- nase (FIST/HIPK3) as a novel Fas-interacting protein. Binding to Fas is mediated by a con- served sequence in the COOH terminus of the protein. FIST/HIPK3 is widely expressed in mammalian tissues and is localized both in the nucleus and in the cytoplasm. In transfected cell lines, FIST/HIPK3 causes FADD phosphorylation, thereby promoting FIST/HIPK3–FADD– Fas interaction. Although Fas ligand–induced activation of Jun NH2-terminal kinase is im- paired by overexpressed active FIST/HIPK3, cell death is not affected. These results suggest that Fas-associated FIST/HIPK3 modulates one of the two major signaling pathways of Fas. Key words: Fas/CD95 • apoptosis • kinase • Jun NH2-terminal kinase • signal transduction Introduction Fas (APO-1/CD95) is a member of the TNFR family and tiate apoptosis by subsequent cleavage of downstream ef- induces apoptosis when cross-linked with Fas ligand (FasL)1 fector caspases (caspase-3, -6, and -7). -

Integrative Genomic Analysis Predicts Causative Cis-Regulatory Mechanisms of the Breast Cancer-Associated Genetic Variant Rs4415084
Author Manuscript Published OnlineFirst on January 19, 2018; DOI: 10.1158/0008-5472.CAN-17-3486 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Cancer Research Mathematical Oncology Title: Integrative genomic analysis predicts causative cis-regulatory mechanisms of the breast cancer-associated genetic variant rs4415084 Authors and affiliations: Yi Zhang1,2*, Mohith Manjunath2*, Shilu Zhang3, Deborah Chasman3, Sushmita Roy3,4, and Jun S. Song2,5 *Equal contribution 1Department of Bioengineering, 2Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. 3Wisconsin Institute for Discovery, 4Department of Biostatistics and Medical Informatics, University of Wisconsin–Madison, Madison, WI 53792, USA. 5Department of Physics, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. Running title: Regulatory genomics of GWAS SNPs Abbreviations: GWAS - genome-wide association study; eQTL - expression quantitative trait loci; ASE - allele-specific expression; TF - transcription factor; LD - linkage disequilibrium; FPKM - fragments per kilobase of transcript per million mapped reads; LCASE - local chromosome allele-specific expression; DHS - DNase I hypersensitive sites; PWM - position weight matrix; TCGA - The Cancer Genome Atlas; ER+ - estrogen receptor positive; TAD - topologically associated domain; MAF - minor allele frequency; RPKM - reads per kilobase of transcript per million mapped reads; SNP - single nucleotide polymorphism; MAPQ - mapping quality; ChIP-seq - chromatin immunoprecipitation sequencing; ASB - allele-specific binding; ncRNA - non-coding RNA; TSS - transcription start site; DNase-seq - DNase I 1 Downloaded from cancerres.aacrjournals.org on September 29, 2021. © 2018 American Association for Cancer Research. Author Manuscript Published OnlineFirst on January 19, 2018; DOI: 10.1158/0008-5472.CAN-17-3486 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -
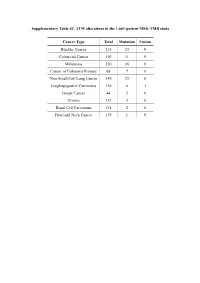
Supplementary Table S1. ATM Alterations in the 1,661-Patient MSK-TMB Study
Supplementary Table S1. ATM alterations in the 1,661-patient MSK-TMB study Cancer Type Total Mutation Fusion Bladder Cancer 215 23 0 Colorectal Cancer 110 11 0 Melanoma 320 26 0 Cancer of Unknown Primary 88 7 0 Non-Small Cell Lung Cancer 350 23 0 Esophagogastric Carcinoma 126 6 1 Breast Cancer 44 2 0 Glioma 117 3 0 Renal Cell Carcinoma 151 2 0 Head and Neck Cancer 139 1 0 Supplementary Table S2. ATM alternations in the 10,945-patient MSK-IMPACT study Cancer Type Total Mutation Deep deletion Amplification Multi-alterations Fusion Small Bowl Cancer 35 7 0 0 0 0 Skin Cancer, Non-Melaoma 148 19 1 0 0 0 Bladder Cancer 423 45 2 0 0 0 Endometrial Cancer 218 24 0 0 0 0 Hepatobiliary Cancer 355 27 0 0 0 0 Colorectal Cancer 1007 75 0 0 0 1 Mature B-Cell Neoplasms 134 8 0 0 2 0 Non-Small Cell Lung Cancer 1668 120 0 2 1 0 Melanoma 365 23 0 0 0 0 Appendiceal Cancer 79 4 0 0 0 0 Small Cell Lung Cancer 82 4 0 0 0 0 Prostate Cancer 717 25 7 1 0 0 Histiocytosis 22 1 0 0 0 0 Salivary Gland Cancer 114 5 0 0 0 0 Thyroid Cancer 231 10 0 0 0 0 Cancer of Unknown Primary 186 8 0 0 0 0 Breast Cancer 1324 48 3 2 0 0 Adrenocortical Carcinoma 25 1 0 0 0 0 Mature T and NK Neoplasms 29 1 0 0 0 0 Renal Cell Carcinoma 361 12 0 0 0 0 Head and Neck Cancer 186 14 0 0 0 1 Pancreatic Cancer 502 69 7 2 0 0 Glioma 553 14 1 0 0 0 Soft Tissue Sarcoma 443 13 1 0 0 0 Esophagogastric Cancer 341 8 1 0 0 0 Peripheral Nervous System 80 0 2 0 0 0 Germ Cell Tumor 288 2 0 1 0 1 Ovarian Can 244 2 1 0 0 0 Uterine Sarcoma 93 1 0 0 0 0 Mesothelioma 107 1 0 0 0 0 Bone Cancer 134 1 0 0 0 0 Gastrointestinal Stromal Ca 137 0 0 0 0 1 Supplementary Table S3. -

Systemic Steroid Exposure Is Associated with Differential Methylation in Chronic Obstructive Pulmonary Disease
Systemic Steroid Exposure Is Associated with Differential Methylation in Chronic Obstructive Pulmonary Disease Emily S. Wan1,2, Weiliang Qiu1, Andrea Baccarelli3, Vincent J. Carey1, Helene Bacherman1, Stephen I. Rennard4, Alvar Agustı´5, Wayne H. Anderson6, David A. Lomas7, and Dawn L. DeMeo1,2 1Channing Division of Network Medicine, Brigham and Women’s Hospital, Boston, Massachusetts; 2Division of Pulmonary and Critical Care Medicine, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts; 3Exposure Epidemiology and Risk Program, Harvard School of Public Health, Boston, Massachusetts; 4Section of Pulmonary and Critical Care, University of Nebraska Medical Center, Omaha, Nebraska; 5Thorax Institute, Hospital Clinic, IDIBAPS, University of Barcelona and CIBER Enfermedades Respiratorias, FISIB, Mallorca, Spain; 6Division of Pulmonary and Critical Care Medicine, University of North Carolina, Chapel Hill, North Carolina; and 7Department of Medicine, Cambridge Institute for Medical Research, University of Cambridge, Cambridge, United Kingdom Rationale: Systemic glucocorticoids are used therapeutically to treat a variety of medical conditions. Epigenetic processes such as DNA AT A GLANCE COMMENTARY methylation may reflect exposure to glucocorticoids and may be involved in mediating the responses and side effects associated Scientific Knowledge on the Subject with these medications. Systemic corticosteroids are associated with a wide range of Objectives: To test the hypothesis that differences in -

Preferentially Paternal Origin of De Novo 11P13 Chromosome Deletions Revealed in Patients with Congenital Aniridia and WAGR Syndrome
G C A T T A C G G C A T genes Communication Preferentially Paternal Origin of De Novo 11p13 Chromosome Deletions Revealed in Patients with Congenital Aniridia and WAGR Syndrome Tatyana A. Vasilyeva 1, Andrey V. Marakhonov 1,* , Natella V. Sukhanova 2, Sergey I. Kutsev 1 and Rena A. Zinchenko 1 1 Research Centre for Medical Genetics, 115522 Moscow, Russia; [email protected] (T.A.V.); [email protected] (S.I.K.); [email protected] (R.A.Z.) 2 Central Clinical Hospital of the Russian Academy of Sciences, 119333 Moscow, Russia; [email protected] * Correspondence: [email protected] Received: 16 June 2020; Accepted: 14 July 2020; Published: 17 July 2020 Abstract: The frequency of pathogenic large chromosome rearrangements detected in patients with different Mendelian diseases is truly diverse and can be remarkably high. Chromosome breaks could arise through different known mechanisms. Congenital PAX6-associated aniridia is a hereditary eye disorder caused by mutations or chromosome rearrangements involving the PAX6 gene. In our recent study, we identified 11p13 chromosome deletions in 30 out of 91 probands with congenital aniridia or WAGR syndrome (characterized by Wilms’ tumor, Aniridia, and Genitourinary abnormalities as well as mental Retardation). The loss of heterozygosity analysis (LOH) was performed in 10 families with de novo chromosome deletion in proband. In 7 out of 8 informative families, the analysis revealed that deletions occurred at the paternal allele. If paternal origin is not random, chromosome breaks could arise either (i) during spermiogenesis, which is possible due to specific male chromatin epigenetic program and its vulnerability to the breakage-causing factors, or (ii) in early zygotes at a time when chromosomes transmitted from different parents still carry epigenetic marks of the origin, which is also possible due to diverse and asymmetric epigenetic reprogramming occurring in male and female pronuclei. -

Transcriptomic and Proteomic Landscape of Mitochondrial
TOOLS AND RESOURCES Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals Inge Ku¨ hl1,2†*, Maria Miranda1†, Ilian Atanassov3, Irina Kuznetsova4,5, Yvonne Hinze3, Arnaud Mourier6, Aleksandra Filipovska4,5, Nils-Go¨ ran Larsson1,7* 1Department of Mitochondrial Biology, Max Planck Institute for Biology of Ageing, Cologne, Germany; 2Department of Cell Biology, Institute of Integrative Biology of the Cell (I2BC) UMR9198, CEA, CNRS, Univ. Paris-Sud, Universite´ Paris-Saclay, Gif- sur-Yvette, France; 3Proteomics Core Facility, Max Planck Institute for Biology of Ageing, Cologne, Germany; 4Harry Perkins Institute of Medical Research, The University of Western Australia, Nedlands, Australia; 5School of Molecular Sciences, The University of Western Australia, Crawley, Australia; 6The Centre National de la Recherche Scientifique, Institut de Biochimie et Ge´ne´tique Cellulaires, Universite´ de Bordeaux, Bordeaux, France; 7Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden Abstract Dysfunction of the oxidative phosphorylation (OXPHOS) system is a major cause of human disease and the cellular consequences are highly complex. Here, we present comparative *For correspondence: analyses of mitochondrial proteomes, cellular transcriptomes and targeted metabolomics of five [email protected] knockout mouse strains deficient in essential factors required for mitochondrial DNA gene (IKu¨ ); expression, leading to OXPHOS dysfunction. Moreover, -

Table SI. List of Differentially Expressed Genes Detected from Three Expression Profiling Datasets GSE18842, GSE30219 and GSE33532
Table SI. List of differentially expressed genes detected from three expression profiling datasets GSE18842, GSE30219 and GSE33532. DEGs Gene Upregulated (n=428) ABCB6, ACP1, ADAM12, ADAMDEC1, ADAMTS12, AFAP1-AS1, AKR1B10, ALDH3B2, ALYREF, ANKRD22, APOBEC3B, ARG2, ARNTL2, ARTN, ASF1B, ATAD2, ATP11B, AUNIP, AURKB, BAIAP2L1, BCL11A, BIK, BIRC5, BLM, BNIP3, BORA, BUB1, BYSL, C10orf2, C12orf56, C12orf66, C15orf48, C2CD4A, C4orf46, C5orf46, CABYR, CALU, CASC7, CBX2, CCNA2, CCNE1, CCNE2, CCT2, CCT6A, CDC25A, CDC25C, CDC45, CDCA2, CDCA3, CDCA8, CDH3, CDK1, CDK5R1, CDKN2A, CDT1, CEMIP, CENPF, CENPH, CENPI, CENPK, CENPL, CENPM, CENPN, MLF1IP, CENPW, CERS6, CHAC2, CHEK1, CIART, CKAP2, CKAP2L, CKAP4, CKS1B, CNFN, COCH, COL11A1, COL3A1, COL5A1, COL5A2, CPD, CRABP2, CST1, CTSV, CTTN, CXCL13, DARS2, DBF4, DCAF13, DCUN1D1, DCUN1D5, DENND1A, DEPDC1, DEPDC1B, DEPDC7, DHFR, DIO2, DONSON, DPY19L1, DSC2, DSCC1, DSG2, DST, DTL, DUS4L, E2F7, ECT2, EGLN3, EIF4EBP1, ELOVL6, ENAH, ENO1, EPT1, ERCC6L, ERO1A, ESPL1, ESRP1, ETV4, EZH2, F12, FAM169A, FAM210A, FAM64A, FAM69A, FAM83A, FAM83D, FAM83F, FAM83H, FANCD2, FANCI, FAP, FAXC, FBXO32, FBXO45, FBXO5, FERMT1, FGF11, FHL2, FLAD1, FNDC1, FOXM1, GAL, GALNT14, GALNT2, GALNT7, GART, GCLC, GCLM, GGH, GINS3, GJB2, GJB6, GMPS, GOLM1, GPX2, GRHL1, GRTP1, GTSE1, H2AFX, HELLS, HILPDA, HIST1H2AE, HIST1H2BC, HIST1H2BH, HIST1H2BJ, HIST1H3F, HIST3H2A, HLTF, HMGA1, HMGA2, HMGB3, HOOK1, HORMAD1, HOXA10, HOXC10, HOXD10, HRASLS, HS6ST2, HSPA4L, HYLS1, IDH2, IGF2BP3, IGFBP2, IGFBP3, IGHM, IGSF9, IL4I1, IQGAP3, IRF6, ITGA11, KCNK1, -

Powerful Gene-Based Testing by Integrating Long-Range Chromatin Interactions and Knockoff Genotypes
medRxiv preprint doi: https://doi.org/10.1101/2021.07.14.21260405; this version posted July 18, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Powerful gene-based testing by integrating long-range chromatin interactions and knockoff genotypes Shiyang Ma1, James L. Dalgleish1, Justin Lee2, Chen Wang1, Linxi Liu3, Richard Gill4;5, Joseph D. Buxbaum6, Wendy Chung7, Hugues Aschard8, Edwin K. Silverman9, Michael H. Cho9, Zihuai He2;10, Iuliana Ionita-Laza1;# 1 Department of Biostatistics, Columbia University, New York, NY, 10032, USA 2 Quantitative Sciences Unit, Department of Medicine, Stanford University, Stanford, CA, 94305, USA 3 Department of Statistics, University of Pittsburgh, Pittsburgh, PA, 15260, USA 4 Department of Human Genetics, Genentech, South San Francisco, CA, 94080, USA 5 Department of Epidemiology, Columbia University, New York, NY, 10032, USA 6 Departments of Psychiatry, Neuroscience, and Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, 10029, USA 7 Department of Pediatrics and Medicine, Herbert Irving Comprehensive Cancer Center, Columbia Uni- versity Irving Medical Center, New York, NY, 10032, USA 8 Department of Computational Biology, Institut Pasteur, Paris, France 9 Channing Division of Network Medicine and Division of Pulmonary and Critical Care Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115, USA 10 Department of Neurology and Neurological Sciences, Stanford University, Stanford, CA 94305, USA # e-mail: [email protected] Abstract Gene-based tests are valuable techniques for identifying genetic factors in complex traits. -

Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity
Diabetes Volume 64, September 2015 3135 Sini Heinonen,1 Jana Buzkova,2 Maheswary Muniandy,1 Risto Kaksonen,1,3 Miina Ollikainen,4 Khadeeja Ismail,4 Antti Hakkarainen,5 Jesse Lundbom,5,6 Nina Lundbom,5 Katriina Vuolteenaho,7 Eeva Moilanen,7 Jaakko Kaprio,8,9,10 Aila Rissanen,1,11 Anu Suomalainen,2,12 and Kirsi H. Pietiläinen1,8,13 Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity Diabetes 2015;64:3135–3145 | DOI: 10.2337/db14-1937 Low mitochondrial number and activity have been sug- and OXPHOS proteins in SAT are downregulated in ac- gested as underlying factors in obesity, type 2 diabetes, quired obesity, and are associated with metabolic distur- and metabolic syndrome. However, the stage at which bances already at the preclinical stage. mitochondrial dysfunction manifests in adipose tissue after the onset of obesity remains unknown. Here we examined subcutaneous adipose tissue (SAT) samples Adipocytes are important contributors to energy balance – from healthy monozygotic twin pairs, 22.8 36.2 years of and metabolic homeostasis. Within these highly dynamic D > 2 age, who were discordant ( BMI 3kg/m, mean length cells, mitochondria are at the center of energy metabo- OBESITY STUDIES 6 n of discordance 6.3 0.3 years, = 26) and concordant lism, using carbohydrates, lipids, and proteins to produce D < 2 n ( BMI 3kg/m, = 14) for body weight, and assessed ATP and metabolites for growth, as well as contributing to their detailed mitochondrial metabolic characteristics: adipocyte differentiation and maturation (1). Mitochon- mitochondrial-related transcriptomes with dysregulated dria possess their own multicopy genome, a 16.6-kb cir- pathways, mitochondrial DNA (mtDNA) amount, mtDNA- cular mitochondrial DNA (mtDNA) that encodes two encoded transcripts, and mitochondrial oxidative phos- phorylation (OXPHOS) protein levels. -

Expression Profile and Bioinformatics Analysis of Circular Rnas in Acute
www.nature.com/scientificreports OPEN Expression profle and bioinformatics analysis of circular RNAs in acute ischemic stroke in a South Chinese Han population Shenghua Li1, Lan Chen2, Chen Xu1, Xiang Qu1, Zhenxiu Qin1, Jinggui Gao1, Jinpin Li1 & Jingli Liu1 ✉ Recent studies have found that circular RNAs (circRNAs) play crucial roles not only in the normal growth and the development of diferent tissues and organs but also in the pathogenesis and progression of various disorders. However, the expression patterns and the function of circRNAs in acute ischemic stroke (AIS) in the South Chinese Han population are unclear. In the present study, RNA sequencing (RNA-seq) data was generated from 3 AIS patients and 3 healthy controls. The circRNAs were detected and identifed by CIRI2 and Find_circ software. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analyses were used to detect the expression of circRNAs. Meanwhile, the potential diagnostic value of the selected circRNAs for AIS was assessed by generating receiver operating characteristic (ROC) curve with area under curve (AUC). The bioinformatic analysis of the host genes of diferentially expressed (DE) circRNAs was performed by gene ontology (GO) enrichment, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, KOBAS for pathway analysis and regulatory network analysis. miRNA-circRNA and miRNA-mRNA interactions were predicted by using TargetScan, miRanda and starBase. CircRNA-miRNA-mRNA interaction networks were created with Cytoscape. Our result showed that there were 2270 DE circRNAs between AIS patients and healthy controls. Among them, 659 were found upregulated and 1611 were downregulated. Bioinformatic analysis showed that the DE circRNAs were related to the following biological processes: endocytosis, energy metabolism, apoptosis, FoxO signaling pathway, platelet activation, neurotrophin signaling pathway and VEGF signaling pathway, which may be associated with the pathological of AIS.