The Freshwater Biological Association
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Toxic Influence of Diazinon As an Organophosphate Pesticide on Parameters of Dry Matter Degradability According to in Situ Technique M
International Journal of Basic & Applied Sciences IJBAS-IJENS Vol:12 No:06 229 Toxic Influence of Diazinon as an Organophosphate Pesticide on Parameters of Dry Matter Degradability According to in Situ Technique M. Kazemi*, A. M. Tahmasbi, R. Valizadeh, A. A. Naserian Department of Animal Science, Excellence Center for Animal Science, Faculty of Agriculture, Ferdowsi University of Mashhad, P. O. Box 91775-1163, Mashhad, Islamic Republic of Iran. *Corresponding author email: [email protected] Abstract-- Many feed and forages are exposed to low, medium metabolizing phosphate insecticides in vitro. Parathion was or high levels of organophosphorous (OP) pesticides. It is unclear reduced to aminoparathion by bovine rumen fluid. Kutches et whether these exposures (for example diazinon as an OP al. [15] reported that toxaphene was the most inhibitory of the pesticide) impact dry matter degradability parameters. So, pesticides tested and resulted in greater decreases of the in different levels (0, 0.7, 2.8 and 5.6 mg) of diazinon as an OP vitro dry matter disappearance, volatile fatty acids and rumen pesticide with different levels of calcium bentonite (CB) (0 and 100 mg) as a toxin binder were tested for their toxicological protozoa. Inhibitory effects of OP pesticides such as effects on in vitro dry matter disappearance. Also we investigated phosphomidon, chlorpyrifos and phosalone on tetrahymena whether CB can inactivate the probably deleterious effects of Spp. Were reported by Hoskin et al. [10]. Dauterman et al. [7] contaminated forage with diazinon in the situ procedure or not. treated rats and cattle with radioactive dimethoate and The study indicated that effect of diazinon with adding the analyzed blood, tissue, milk, and excreta. -
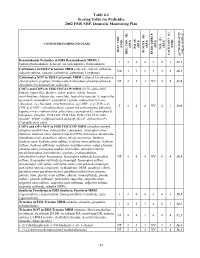
2002 NRP Section 6, Tables 6.1 Through
Table 6.1 Scoring Table for Pesticides 2002 FSIS NRP, Domestic Monitoring Plan } +1 0.05] COMPOUND/COMPOUND CLASS * ) (EPA) (EPA) (EPA) (EPA) (EPA) (FSIS) (FSIS) PSI (P) TOX.(T) L-1 HIST. VIOL. BIOCON. (B) {[( (2*R+P+B)/4]*T} REG. CON. (R) * ENDO. DISRUP. LACK INFO. (L) LACK INFO. {[ Benzimidazole Pesticides in FSIS Benzimidazole MRM (5- 131434312.1 hydroxythiabendazole, benomyl (as carbendazim), thiabendazole) Carbamates in FSIS Carbamate MRM (aldicarb, aldicarb sulfoxide, NA44234416.1 aldicarb sulfone, carbaryl, carbofuran, carbofuran 3-hydroxy) Carbamates NOT in FSIS Carbamate MRM (carbaryl 5,6-dihydroxy, chlorpropham, propham, thiobencarb, 4-chlorobenzylmethylsulfone,4- NT 4 1 3 NV 4 4 13.8 chlorobenzylmethylsulfone sulfoxide) CHC's and COP's in FSIS CHC/COP MRM (HCB, alpha-BHC, lindane, heptachlor, dieldrin, aldrin, endrin, ronnel, linuron, oxychlordane, chlorpyrifos, nonachlor, heptachlor epoxide A, heptachlor epoxide B, endosulfan I, endosulfan I sulfate, endosulfan II, trans- chlordane, cis-chlordane, chlorfenvinphos, p,p'-DDE, p, p'-TDE, o,p'- 3444NV4116.0 DDT, p,p'-DDT, carbophenothion, captan, tetrachlorvinphos [stirofos], kepone, mirex, methoxychlor, phosalone, coumaphos-O, coumaphos-S, toxaphene, famphur, PCB 1242, PCB 1248, PCB 1254, PCB 1260, dicofol*, PBBs*, polybrominated diphenyl ethers*, deltamethrin*) (*identification only) COP's and OP's NOT in FSIS CHC/COP MRM (azinphos-methyl, azinphos-methyl oxon, chlorpyrifos, coumaphos, coumaphos oxon, diazinon, diazinon oxon, diazinon met G-27550, dichlorvos, dimethoate, dimethoate -

Risk Mitigation Methods for Removal of Pesticide Residues in Brinjal for Food Safety
Universal Journal of Agricultural Research 2(8): 279-283, 2014 http://www.hrpub.org DOI: 10.13189/ujar.2014.020801 Risk Mitigation Methods for Removal of Pesticide Residues in Brinjal for Food Safety Cherukuri Sreenivasa Rao*, Vemuri Shashi Bhushan, Harinatha Reddy A. , Ravindranath Darsi, Aruna M. , Ramesh B. Professor Jayashankar Telangana State Agricultural University, Rajendranagar, Hyderabad-500030, Andhra Pradesh, India *Corresponding Author: [email protected] Copyright © 2014 Horizon Research Publishing All rights reserved. Abstract The commercial production of highly 1. Introduction cultivated and consumed brinjal is highly dependent on regular usage of insecticides to protect the crop from insect Brinjal is the most popular vegetable in India, and state of pests. The increased consumer awareness and legal issues on Andhra Pradesh is third most important growing Brinjal food safety, with special reference to insecticide residues in producing 1.615 M mt with a share of 12% (NHB, 2013) foods, led us to attempt for cheap and effective methods for during 2012-13. In India, about 13-14% of the total removal of pesticide residues to address the issues of pesticides used in agriculture are used for fruits and consumer and food safety, as the farmers are not following vegetables covering only 3% of the cropped area. Repeated the Good Agricultural Practices i.e pre-harvest intervals. The application of pesticides on vegetables often results in the most commonly used pesticides such as profenophos, buildup of their residues. Surveys carried out in the country chlorpyriphos, dimethoate, malathion, phosalone, indicated that 50-70% of vegetables are contaminated with quinalphos, triazophos and -cyhalothrin were sprayed at insecticide residues).Studies on farm gate monitoring of recommended doses at brinjal formation stage, samples were vegetables carried out in different places revealed collected at 2 hours after treatment to quantify the deposits. -

Environmental Health Criteria 63 ORGANOPHOSPHORUS
Environmental Health Criteria 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION Please note that the layout and pagination of this web version are not identical with the printed version. Organophophorus insecticides: a general introduction (EHC 63, 1986) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1986 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination -

Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 Theinternational Programme on Chemical Safety (IPCS) Was Established in 1980
The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 cation Hazard of Pesticides by and Guidelines to Classi The WHO Recommended Classi The WHO Recommended Classi cation of Pesticides by Hazard and Guidelines to Classi cation 2019 The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification 2019 TheInternational Programme on Chemical Safety (IPCS) was established in 1980. The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. This publication was developed in the IOMC context. The contents do not necessarily reflect the views or stated policies of individual IOMC Participating Organizations. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase international coordination in the field of chemical safety. The Participating Organizations are: FAO, ILO, UNDP, UNEP, UNIDO, UNITAR, WHO, World Bank and OECD. The purpose of the IOMC is to promote coordination of the policies and activities pursued by the Participating Organizations, jointly or separately, to achieve the sound management of chemicals in relation to human health and the environment. WHO recommended classification of pesticides by hazard and guidelines to classification, 2019 edition ISBN 978-92-4-000566-2 (electronic version) ISBN 978-92-4-000567-9 (print version) ISSN 1684-1042 © World Health Organization 2020 Some rights reserved. -

Pesticide Residues : Maximum Residue Limits
THAI AGRICULTURAL STANDARD TAS 9002-2013 PESTICIDE RESIDUES : MAXIMUM RESIDUE LIMITS National Bureau of Agricultural Commodity and Food Standards Ministry of Agriculture and Cooperatives ICS 67.040 ISBN UNOFFICAL TRANSLATION THAI AGRICULTURAL STANDARD TAS 9002-2013 PESTICIDE RESIDUES : MAXIMUM RESIDUE LIMITS National Bureau of Agricultural Commodity and Food Standards Ministry of Agriculture and Cooperatives 50 Phaholyothin Road, Ladyao, Chatuchak, Bangkok 10900 Telephone (662) 561 2277 Fascimile: (662) 561 3357 www.acfs.go.th Published in the Royal Gazette, Announcement and General Publication Volume 131, Special Section 32ง (Ngo), Dated 13 February B.E. 2557 (2014) (2) Technical Committee on the Elaboration of the Thai Agricultural Standard on Maximum Residue Limits for Pesticide 1. Mrs. Manthana Milne Chairperson Department of Agriculture 2. Mrs. Thanida Harintharanon Member Department of Livestock Development 3. Mrs. Kanokporn Atisook Member Department of Medical Sciences, Ministry of Public Health 4. Mrs. Chuensuke Methakulawat Member Office of the Consumer Protection Board, The Prime Minister’s Office 5. Ms. Warunee Sensupa Member Food and Drug Administration, Ministry of Public Health 6. Mr. Thammanoon Kaewkhongkha Member Office of Agricultural Regulation, Department of Agriculture 7. Mr. Pisan Pongsapitch Member National Bureau of Agricultural Commodity and Food Standards 8. Ms. Wipa Thangnipon Member Office of Agricultural Production Science Research and Development, Department of Agriculture 9. Ms. Pojjanee Paniangvait Member Board of Trade of Thailand 10. Mr. Charoen Kaowsuksai Member Food Processing Industry Club, Federation of Thai Industries 11. Ms. Natchaya Chumsawat Member Thai Agro Business Association 12. Mr. Sinchai Swasdichai Member Thai Crop Protection Association 13. Mrs. Nuansri Tayaputch Member Expert on Method of Analysis 14. -

Target Pesticide List
EUPT-FV17- TARGET PESTICIDE LIST MRRL Pesticide (mg/Kg) 3-hydroxy-carbofuran 0.01 Acephate 0.01 Acetamiprid 0.01 Acrinathrin 0.01 Aldicarb 0.01 Aldicarb Sulfone 0.01 Aldicarb Sulfoxide 0.01 Azinphos-methyl 0.01 Azoxystrobin 0.01 Benfuracarb 0.01 Benomyl 0.01 Bifenthrin 0.01 Bitertanol 0.01 Boscalid 0.01 Bromopropylate 0.01 Bromuconazole 0.01 Bupirimate 0.01 Buprofezin 0.01 Cadusafos 0.006 Carbaryl 0.01 Carbendazim 0.01 Carbofuran 0.01 Carbosulfan 0.01 Chlorfenapyr 0.01 Chlorfenvinphos 0.01 Chlorobenzilate 0.01 Chlorothalonil 0.01 Chlorpropham (only parent compound) 0.01 Chlorpyrifos 0.01 Chlorpyrifos-methyl 0.01 Clofentezine (only parent compound) 0.01 Clothianidin 0.01 Cyfluthrin (cyfluthrin incl. other mixtures of constituent isomers (sum of isomers)) 0.01 Cypermethrin (cypermethrin incl. other mixtures of constituent isomers (sum of isomers)) 0.01 Cyproconazole 0.01 Cyprodinil 0.01 Deltamethrin 0.01 Demeton-S-methylsulfone 0.006 Desmethyl-pirimicarb 0.01 Diazinon 0.01 Dichlofluanid (only parent compound) 0.01 Dichlorvos 0.01 Dicloran 0.01 Dicofol 0.01 Diethofencarb 0.01 Difenoconazole 0.01 Diflubenzuron 0.01 Dimethoate 0.003 Dimethomorph 0.01 Dimethylaminosulfotoluidide (DMST) 0.01 Diniconazole 0.01 Diphenylamine 0.01 Endosulfan alpha 0.01 Endosulfan beta 0.01 Endosulfan sulfate 0.01 EPN 0.01 Epoxiconazole 0.01 Ethion 0.01 Ethirimol 0.01 Ethoprophos 0.008 Etofenprox 0.01 Fenamidone 0.01 Fenamiphos 0.01 Fenamiphos sulfone 0.01 Fenamiphos sulfoxide 0.01 Fenarimol 0.01 Fenazaquin 0.01 Fenbuconazole 0.01 Fenhexamid 0.01 Fenitrothion 0.01 -
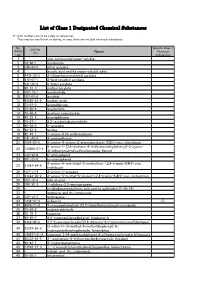
List of Class 1 Designated Chemical Substances
List of Class 1 Designated Chemical Substances *1:CAS numbers are to be solely as references. They may be insufficient or lacking, in case there are multiple chemical substances. No. Specific Class 1 CAS No. (PRTR Chemical (*1) Name Law) Substances 1 - zinc compounds(water-soluble) 2 79-06-1 acrylamide 3 140-88-5 ethyl acrylate 4 - acrylic acid and its water-soluble salts 5 2439-35-2 2-(dimethylamino)ethyl acrylate 6 818-61-1 2-hydroxyethyl acrylate 7 141-32-2 n-butyl acrylate 8 96-33-3 methyl acrylate 9 107-13-1 acrylonitrile 10 107-02-8 acrolein 11 26628-22-8 sodium azide 12 75-07-0 acetaldehyde 13 75-05-8 acetonitrile 14 75-86-5 acetone cyanohydrin 15 83-32-9 acenaphthene 16 78-67-1 2,2'-azobisisobutyronitrile 17 90-04-0 o-anisidine 18 62-53-3 aniline 19 82-45-1 1-amino-9,10-anthraquinone 20 141-43-5 2-aminoethanol 21 1698-60-8 5-amino-4-chloro-2-phenylpyridazin-3(2H)-one; chloridazon 5-amino-1-[2,6-dichloro-4-(trifluoromethyl)phenyl]-3-cyano- 22 120068-37-3 4[(trifluoromethyl)sulfinyl]pyrazole; fipronil 23 123-30-8 p-aminophenol 24 591-27-5 m-aminophenol 4-amino-6-tert-butyl-3-methylthio-1,2,4-triazin-5(4H)-one; 25 21087-64-9 metribuzin 26 107-11-9 3-amino-1-propene 27 41394-05-2 4-amino-3-methyl-6-phenyl-1,2,4-triazin-5(4H)-one; metamitron 28 107-18-6 allyl alcohol 29 106-92-3 1-allyloxy-2,3-epoxypropane 30 - n-alkylbenzenesulfonic acid and its salts(alkyl C=10-14) 31 - antimony and its compounds 32 120-12-7 anthracene 33 1332-21-4 asbestos ○ 34 4098-71-9 3-isocyanatomethyl-3,5,5-trimethylcyclohexyl isocyanate 35 78-84-2 isobutyraldehyde -

NMP-Free Formulations of Neonicotinoids
(19) & (11) EP 2 266 400 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 29.12.2010 Bulletin 2010/52 A01N 43/40 (2006.01) A01N 43/86 (2006.01) A01N 47/40 (2006.01) A01N 51/00 (2006.01) (2006.01) (2006.01) (21) Application number: 09305544.0 A01P 7/00 A01N 25/02 (22) Date of filing: 15.06.2009 (84) Designated Contracting States: (72) Inventors: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR • Gasse, Jean-Jacques HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL 27600 Saint-Aubin-Sur-Gaillon (FR) PT RO SE SI SK TR • Duchamp, Guillaume Designated Extension States: 92230 Gennevilliers (FR) AL BA RS • Cantero, Maria 92230 Gennevilliers (FR) (71) Applicant: NUFARM 92233 Gennevelliers (FR) (74) Representative: Cabinet Plasseraud 52, rue de la Victoire 75440 Paris Cedex 09 (FR) (54) NMP-free formulations of neonicotinoids (57) The invention relates to NMP-free liquid formulation comprising at least one nicotinoid and at least one aprotic polar component selected from the group comprising the compounds of formula I, II or III below, and mixtures thereof, wherein R1 and R2 independently represent H or an alkyl group having less than 5 carbons, preferably a methyl group, and n represents an integer ranging from 0 to 5, and to their applications. EP 2 266 400 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 266 400 A1 Description Technical Field of the invention 5 [0001] The invention relates to novel liquid formulations of neonicotinoids and to their use for treating plants, for protecting plants from pests and/or for controlling pests infestation. -

Compounds of Potential Interest
TABLE A-1 INFORMATION ON COMPOUNDS OF POTENTIAL INTEREST (Page 1 of 24) CAS Number Compound Name Detected U.S. EPA Identified in PICs in Stack Data Available for All HHRAs Emissions (1993) 40 CFR Part 261 Combustion Unit Chemical-Specific Recommended and Emissions Actually PICs Recommended by U.S. EPA (1994c) Potential PICs (1994) Compounds Listed in Appendix VII or VIII U.S. EPA Compounds 50-00-0 Formaldehyde (methylene oxide) K009, K010, K038, K040, K156, K157 X X X 50-06-6 Phenobarbital 50-07-7 Mitomycin 50-18-0 Cyclophosphamide 50-29-3 4,4'-DDT X F032, F034, F037, F038, K001, K022, K035, K141, K142, 50-32-8 Benzo(a)pyrene X X X X K144, K145, K147, K148 50-55-5 Reserpine 51-28-5 2,4-Dinitrophenol K001 X X 51-43-4 Epinephrine 51-52-5 Propylthiouracil 51-79-6 Ethyl carbamate (urethane) X 52-85-7 Famphur 53-70-3 Dibenzo(a,h)anthracene F032, F034, K022, K141, K142, K144, K145, K147, K148 X X 53-96-3 2-Acetylaminofluorene 54-11-5 Nicotine 55-18-5 Nitrosodiethylamine 55-38-9 Fenthion 55-63-0 Nitroglycerine 55-91-4 Diisopropylfluorophosphate (DFP) 56-04-2 Methylthiouracil F001, F024, F025, K016, K019, K020, K021, K073, K116, 56-23-5 Carbon tetrachloride X X X X K150, K151, K157 56-38-2 Parathion 56-49-5 3-Methylcholanthrene 56-53-1 Diethylstilbestrol F032, F034, K001, K022, K035, K141, K142, K143, K144, 56-55-3 Benzo(a)anthracene X X X K145, K147, K148 56-57-5 Nitroquinoline-1-oxide Note: See Table A-1 References and Discussion (Appendix A-1) for explanation of the information presented. -

522 PART 185—TOLERANCES for PESTICIDES in FOOD Subpart A
Pt. 185 40 CFR Ch. I (7±1±98 Edition) peaches, pears, strawberries, and toma- 185.3550 Hexakis. toes. This temporary exemption from 185.3575 Hexazinone. the requirement for a tolerance will 185.3600 Hydrogen cyanide. 185.3650 Imazalil. permit the marketing of the food com- 185.3700 Inorganic bromide. modities in this paragraph when treat- 185.3750 Iprodione. ed in accordance with the provisions of 185.3775 d-Limonene. experimental use permit 70515-EUP-1, 185.3800 Magnesium phosphide. which is being issued under the Federal 185.4000 Metalaxyl. Insecticide, Fungicide, and Rodenticide 185.4035 Metarhizium anisopliae strain Act (FIFRA), as amended (7 U.S.C. 136). ESF1. 185.4100 Methomyl. This temporary exemption from the re- 185.4150 Methoprene. quirement of a tolerance expires and is 185.4250 Methyl chloride. revoked on June 1, 2001. This tem- 185.4300 Methyl formate. porary exemption from the require- 185.4400 Nitrogen. ment of a tolerance may be revoked at 185.4500 N-Octylbicycloheptene any time if the experimental use per- dicarboximide. 185.4650 Paraformaldehyde. mit is revoked or if any experience 185.4800 Phosalone. with or scientific data on this pesticide 185.4850 Picloram. indicate that the tolerance is not safe. 185.4900 Piperonyl butoxide. 185.4950 Pirimiphos-methyl. [63 FR 32134, June 12, 1998] 185.5000 Propargite. 185.5100 Propetamphos. PART 185ÐTOLERANCES FOR 185.5150 Propylene oxide. PESTICIDES IN FOOD 185.5200 Pyrethrins. 185.5375 Sulfonium, trimethyl-salt with N- (phosphonomethyl)glycine (1:1). Subpart A [Reserved] 185.5950 Triforine. 185.6300 Zinc ion and maneb coordination Subpart BÐFood Additives Permitted in product. -
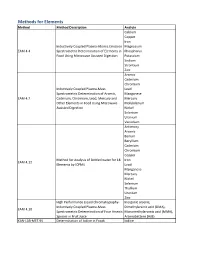
Method Description
Methods for Elements Method Method Description Analyte Calcium Copper Iron Inductively Coupled Plasma-Atomic Emission Magnesium EAM 4.4 Spectrometric Determination of Elements in Phosphorus Food Using Microwave Assisted Digestion Potassium Sodium Strontium Zinc Arsenic Cadmium Chromium Inductively Coupled Plasma-Mass Lead Spectrometric Determination of Arsenic, Manganese EAM 4.7 Cadmium, Chromium, Lead, Mercury and Mercury Other Elements in Food Using Microwave Molybdenum Assisted Digestion Nickel Selenium Uranium Vanadium Antimony Arsenic Barium Beryllium Cadmium Chromium Copper Method for Analysis of Bottled water for 18 Iron EAM 4.12 Elements by ICPMS Lead Manganese Mercury Nickel Selenium Thallium Uranium Zinc High Performance Liquid Chromatography- Inorganic arsenic, Inductively Coupled Plasma-Mass Dimethylarsinic acid (DMA), EAM 4.10 Spectrometric Determination of Four Arsenic Monomethylarsonic acid (MMA), Species in Fruit Juice Arsenobetaine (AsB) KAN-LAB-MET.95 Determination of Iodine in Foods Iodine Methods for Radionuclides Method Method Description Analyte Determination of Strontium-90 in Foods by WEAC.RN.METHOD.2.0 Strontium-90 Internal Gas-Flow Proportional Counting Americium-241 Cesium-134 Cesium-137 Determination of Gamma-Ray Emitting Cobalt-60 WEAC.RN.METHOD.3.0 Radionuclides in Foods by High-Purity Potassium-40 Germanium Spectrometry Radium-226 Ruthenium-103 Ruthenium-106 Thorium-232 Methods for Pesticides/Industrial Chemicals Method Method Description Analyte Extraction Method: Analysis of Pesticides KAN-LAB-PES.53 and