Copolymers of Ethylene and 1,3-Butadiene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SAFETY DATA SHEET Cyclopentane
SAFETY DATA SHEET Cyclopentane Section 1. Identification GHS product identifier : Cyclopentane Chemical name : cyclopentane Other means of : cyclopentane (dot); pentamethylene identification Product use : Synthetic/Analytical chemistry. Synonym : cyclopentane (dot); pentamethylene SDS # : 001124 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 2 substance or mixture AQUATIC HAZARD (ACUTE) - Category 3 AQUATIC HAZARD (LONG-TERM) - Category 3 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Highly flammable liquid and vapor. May form explosive mixtures in Air. Harmful to aquatic life with long lasting effects. Precautionary statements General : Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Prevention : Wear protective gloves. Wear eye or face protection. Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. No smoking. Use explosion- proof electrical, ventilating, lighting and all material-handling equipment. Use only non- sparking tools. Take precautionary measures against static discharge. Keep container tightly closed. Avoid release to the environment. Response : IF ON SKIN (or hair): Take off immediately all contaminated clothing. Rinse skin with water or shower. Storage : Store in a well-ventilated place. Keep cool. Disposal : Dispose of contents and container in accordance with all local, regional, national and international regulations. Hazards not otherwise : None known. -
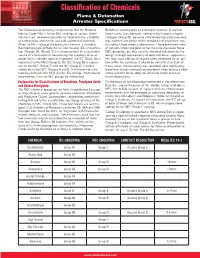
Classification of Chemicals
Classification of Chemicals Flame & Detonation Arrester Specifications PROTECTOSEAL ® The Protectoseal Company recommends that the National Butadiene would qualify as a Group D material. In each of Electric Code (NEC) Article 500, rankings of various chemi - these cases, the chemicals were primarly listed in a higher cals be used, whenever possible, to determine the suitability category (Group B), because of relatively high pressure read - of a detonation arrester for use with a particular chemical. ings noted in one phase of the standard test procedure con - When no NEC rating of the particular chemical is available, ducted by Underwriters Laboratories. These pressures were the International Electrotechnical Commission (IEC) classifica - of concern when categorizing the chemicals because these tion (Groups IIA, IIB and IIC) is recommended as a secondary NEC groupings are also used as standard indicators for the source of information for determining the suitability of an ar - design strength requirements of electrical boxes, apparatus, rester for its intended service. In general, the IEC Group IIA is etc. that must withstand the pressures generated by an igni - equivalent to the NEC Group D; the IEC Group IIB is equiva - tion within the container. It should be noted that, in each of lent to the NEC Group C; and the IEC Group IIC includes these cases, the test pressures recorded were significantly chemicals in the NEC Groups A and B. In the event of a dis - lower than those commonly encountered when testing a deto - crepancy between the NEC and the IEC ratings, Protectoseal nation arrester for its ability to withstand stable and over - recommends that the NEC groups be referenced. -

MSDS Cyclopentane (1).Pdf
MATERIAL SAFETY DATA SHEET (MSDS) CYCLOPENTANE 1. CHEMICAL PRODUCT AND COMPANY IDENTIFICATION SUBSTANCE OR PREPATION TRADE NAME Cyclopentane CHEMICAL CLASSIFICATION Cycloparaffin, Naphthene COMPANY/ UNDERTAKING NAME AND Haldia Petrochemicals Limited, ADDRESS PO Box No 12, Haldia Plant PO Durgachak, Dist Midnapore West Bengal, India PIN 721 602 TELEPHONE 091-3224-274384 / 274400 EMERGENCY TELEPHONE NUMBER 091-3224-275916 2. COMPOSTION AND INFORMATION ON INGREDIENTS CHEMICAL CHEMICLAL CONTENT CAS EXPOSURE LIMITS IN AIR NAME FORMULA NUMBER (ppm) ACGIH ACGIH IDLH TLV- TLV- TWA STEL Cyclopentane C5H10 98.20 wt% 287-92-3 600 NA NA 2,2 Dimethyl (CH3)3CCH2CH3 0.28 wt% 75-83-2 500 1000 NA butane n- pentane n- C5H12 1.47 wt% 109-66-0 600 750 NA i- Pentane i- C5H12 0.05 wt% 78-78-4 600 750 NA Total Sulpher S < 1.0 wt. ppm 7704-34-9 NA NA NA 3. HAZARD CLASSIFICATION Highly flammable liquid and vapour. Vapour EMERGENCY OVERVIEW may cause flash fire. Harmful or Fatal is enter lungs and cause damage POTENTIAL HEALTH HAZARDS EYE SKIN INHALATION INGESTION OTHERS ACUTE To cause Not expected Dizziness, Abdominal prolonged or to be harmful headache, pain, significant to internal nausea, diarrhoea, eye irritation organs if Unconsciousness, dizziness, absorbed weakness nausea, sore through the throat skin. CHRONIC Repeated or prolonged contact with skin may cause dermatitis NFPA HAZARD HEALTH FLAMMABILITY REACTIVITY SPECIAL SIGNALS 2 3 0 - HAZCHEM CODE 3YE GHS-Classification Flammable liquids Category 2 Aspiration hazard Category 1 Chronic aquatic toxicity, Category 3 Acute aquatic toxicity, Category 3 Target Organ Systemic Toxicant - Single exposure, Category 3, Inhalation, Central nervous system Document Compiled By Approved By Issue No Rev. -

Cycloalkanes, Cycloalkenes, and Cycloalkynes
CYCLOALKANES, CYCLOALKENES, AND CYCLOALKYNES any important hydrocarbons, known as cycloalkanes, contain rings of carbon atoms linked together by single bonds. The simple cycloalkanes of formula (CH,), make up a particularly important homologous series in which the chemical properties change in a much more dramatic way with increasing n than do those of the acyclic hydrocarbons CH,(CH,),,-,H. The cyclo- alkanes with small rings (n = 3-6) are of special interest in exhibiting chemical properties intermediate between those of alkanes and alkenes. In this chapter we will show how this behavior can be explained in terms of angle strain and steric hindrance, concepts that have been introduced previously and will be used with increasing frequency as we proceed further. We also discuss the conformations of cycloalkanes, especially cyclo- hexane, in detail because of their importance to the chemistry of many kinds of naturally occurring organic compounds. Some attention also will be paid to polycyclic compounds, substances with more than one ring, and to cyclo- alkenes and cycloalkynes. 12-1 NOMENCLATURE AND PHYSICAL PROPERTIES OF CYCLOALKANES The IUPAC system for naming cycloalkanes and cycloalkenes was presented in some detail in Sections 3-2 and 3-3, and you may wish to review that ma- terial before proceeding further. Additional procedures are required for naming 446 12 Cycloalkanes, Cycloalkenes, and Cycloalkynes Table 12-1 Physical Properties of Alkanes and Cycloalkanes Density, Compounds Bp, "C Mp, "C diO,g ml-' propane cyclopropane butane cyclobutane pentane cyclopentane hexane cyclohexane heptane cycloheptane octane cyclooctane nonane cyclononane "At -40". bUnder pressure. polycyclic compounds, which have rings with common carbons, and these will be discussed later in this chapter. -

Cyclopentane Cyp
CYCLOPENTANE CYP CAUTIONARY RESPONSE INFORMATION 4. FIRE HAZARDS 7. SHIPPING INFORMATION 4.1 Flash Point: 7.1 Grades of Purity: Commercial; 60% (remainder Common Synonyms Watery liquid Colorless Mild, sweet odor < 20°F C.C. consists of hydrocarbons of similar boiling Pentamethylene 4.2 Flammable Limits in Air: (approx.) 1.1%- point); Research: 99+% 8.7% 7.2 Storage Temperature: Ambient Floats on water. Flammable, irritating vapor is produced. 4.3 Fire Extinguishing Agents: Dry 7.3 Inert Atmosphere: No requirement chemical, foam, carbon dioxide Keep people away. 7.4 Venting: Pressure-vacuum Avoid inhalation. 4.4 Fire Extinguishing Agents Not to Be Shut off ingition sources. Call fire department. Used: Water may be ineffective. 7.5 IMO Pollution Category: (C) Evacuate area in case of large discharge. 4.5 Special Hazards of Combustion 7.6 Ship Type: 3 Notify local health and pollution control agencies. Products: Not pertinent 7.7 Barge Hull Type: Currently not available Protect water intakes. 4.6 Behavior in Fire: Containers may explode. FLAMMABLE. 8. HAZARD CLASSIFICATIONS Fire 4.7 Auto Ignition Temperature: 682°F Containers may explode in fire. 8.1 49 CFR Category: Flammable liquid Flashback along vapor trail may occur. 4.8 Electrical Hazards: Not pertinent 8.2 49 CFR Class: 3 Vapor may explode if ignited in an enclosed area. 4.9 Burning Rate: 7.9 mm/min. 8.3 49 CFR Package Group: II Extinguish with dry chemicals, foam or carbon dioxide. 4.10 Adiabatic Flame Temperature: Currently Water may be ineffective on fire. not available 8.4 Marine Pollutant: No Cool exposed containers with water. -

Section 2. Hazards Identification OSHA/HCS Status : This Material Is Considered Hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200)
SAFETY DATA SHEET Flammable Liquefied Gas Mixture: 2-Methylpentane / 2-Methylhexane / 2, 3-Dimethylheptane / Benzene / Cyclohexane / Cyclopentane / Dodecane / Ethyl Benzene / Ethyl Cyclohexane / Hexane / Isobutane / Isobutyl Cyclohexane / Isooctane / Isopentane / Isopropylcyclohexane / Methyl Cyclohexane / N-Butane / N-Butyl Benzene / N-Decane / N-Heptane / N-Nonane / N-Octane / N-Pentane / N-Propyl Benzene / Toluene Section 1. Identification GHS product identifier : Flammable Liquefied Gas Mixture: 2-Methylpentane / 2-Methylhexane / 2, 3-Dimethylheptane / Benzene / Cyclohexane / Cyclopentane / Dodecane / Ethyl Benzene / Ethyl Cyclohexane / Hexane / Isobutane / Isobutyl Cyclohexane / Isooctane / Isopentane / Isopropylcyclohexane / Methyl Cyclohexane / N-Butane / N-Butyl Benzene / N-Decane / N-Heptane / N-Nonane / N-Octane / N-Pentane / N-Propyl Benzene / Toluene Other means of : Not available. identification Product type : Liquefied gas Product use : Synthetic/Analytical chemistry. SDS # : 021986 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE GASES - Category 1 substance or mixture GASES UNDER PRESSURE - Liquefied gas SKIN IRRITATION - Category 2 GERM CELL MUTAGENICITY - Category 1 CARCINOGENICITY - Category 1 TOXIC TO REPRODUCTION (Fertility) - Category 2 TOXIC TO REPRODUCTION (Unborn child) - Category 2 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (REPEATED EXPOSURE) - Category 1 AQUATIC HAZARD (ACUTE) - Category 2 AQUATIC HAZARD (LONG-TERM) - Category 2 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable gas. May form explosive mixtures with air. -

SAFETY DATA SHEET Cyclopentane
SAFETY DATA SHEET Based upon Regulation (EC) No. 1907/2006, as amended by Regulation (EC) No. 453/2010 cyclopentane SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1 Product identifier: Product name : cyclopentane Synonyms : pentamethylene Registration number REACH : 01-2119463053-47 Product type REACH : Substance/mono-constituent CAS number : 287-92-3 EC index number : 601-030-00-2 EC number : 206-016-6 RTECS number : GY2390000 Molecular mass : 70.14 g/mol Formula : C5H10 1.2 Relevant identified uses of the substance or mixture and uses advised against: 1.2.1 Relevant identified uses Industrial and professional use. Before use: carry out a risk assessment 1.2.2 Uses advised against No uses advised against known 1.3 Details of the supplier of the safety data sheet: Supplier of the safety data sheet Distributor of the product 1.4 Emergency telephone number: 24h/24h (Telephone advice: English, French, German, Dutch): +32 14 58 45 45 (BIG) SECTION 2: Hazards identification 2.1 Classification of the substance or mixture: Classified as dangerous according to the criteria of Regulation (EC) No 1272/2008 Class Category Hazard statements Flam. Liq. category 2 H225: Highly flammable liquid and vapour. Aquatic Chronic category 3 H412: Harmful to aquatic life with long lasting effects. 2.2 Label elements: Signal word Danger H-statements H225 Highly flammable liquid and vapour. H412 Harmful to aquatic life with long lasting effects. P-statements P210 Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. No smoking. P280 Wear protective gloves and eye protection/face protection. -

S-SBR Solution Styrene-Butadiene Rubber Copolymers Versalis Proprietary Process Technologies Available for Licensing
Proprietary process technology S-SBR Solution Styrene-Butadiene Rubber copolymers Versalis proprietary process technologies available for licensing II 1 Our company Our commitment to excellence, in quality of our Versalis – the petrochemical subsidiary of Eni – is products and services, makes our company an active a dynamic player in its industry sector facing the partner for the growth of customers involved in multifold market needs through different skills. petrochemical business. With a history as European manufacturer with more Through engineering services, technical assistance, than 50 years of operating experience, Versalis stands marketing support and continuous innovation, our as a complete, reliable and now global supplier in the knowledge is the key strength to customize any new basic chemicals, intermediates, plastics and elastomers project throughout all phases. market with a widespread sales network. Customers can rely on this strong service-oriented Relying on continuous development in its production outlook and benefit from a product portfolio that plants as well as in its products, strengthening the strikes a perfect balance of processability and management of the knowledge gained through its long mechanical properties, performance and industrial experience, Versalis has become a worldwide eco-friendliness. licensor of its proprietary technologies and proprietary catalysts. The strong integration between R&D, Technology and Engineering departments, as well as a deep market expertise, are the key strengths for finding answers -

Section 13. Disposal Considerations Disposal Methods : the Generation of Waste Should Be Avoided Or Minimized Wherever Possible
SAFETY DATA SHEET Flammable Liquid Mixture: 2-Methylpentane / 2,3-Dimethylbutane / 3-Methylpentane / Benzene / Cyclohexane / Cyclopentane / Hexane / Isopentane / Methyl Cyclopentane / N-Butane / N-Pentane Section 1. Identification GHS product identifier : Flammable Liquid Mixture: 2-Methylpentane / 2,3-Dimethylbutane / 3-Methylpentane / Benzene / Cyclohexane / Cyclopentane / Hexane / Isopentane / Methyl Cyclopentane / N-Butane / N-Pentane Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 019890 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 1 substance or mixture SKIN CORROSION/IRRITATION - Category 2 GERM CELL MUTAGENICITY - Category 1B CARCINOGENICITY - Category 1 TOXIC TO REPRODUCTION (Fertility) - Category 2 TOXIC TO REPRODUCTION (Unborn child) - Category 2 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (REPEATED EXPOSURE) - Category 2 AQUATIC HAZARD (ACUTE) - Category 3 AQUATIC HAZARD (LONG-TERM) - Category 2 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable liquid and vapor. May form explosive mixtures in Air. Causes skin irritation. May cause genetic defects. May cause cancer. Suspected of damaging fertility or the unborn child. May cause drowsiness and dizziness. May cause damage to organs through prolonged or repeated exposure. Toxic to aquatic life with long lasting effects. Precautionary statements General : Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. -

Supplement of OH and HO2 Radical Chemistry in a Midlatitude Forest: Measurements and Model Comparisons
Supplement of Atmos. Chem. Phys., 20, 9209–9230, 2020 https://doi.org/10.5194/acp-20-9209-2020-supplement © Author(s) 2020. This work is distributed under the Creative Commons Attribution 4.0 License. Supplement of OH and HO2 radical chemistry in a midlatitude forest: measurements and model comparisons Michelle M. Lew et al. Correspondence to: Philip S. Stevens ([email protected]) The copyright of individual parts of the supplement might differ from the CC BY 4.0 License. Table S1: Characterization of measured compounds by various institutions (IU: Indiana University, UMass: University of Massachusetts, LD: IMT Lille Douai) into RACM2 groups. RACM2 VOCs Source RACM2 VOCs Source NO NO UMass HCHO Formaldehyde LD NO2 UMass Acetaldehyde LD NO2 ACD Water vapor IU Acetone LD H2O ACT HONO Nitrous Acid IU EOH Ethanol LD ACE Acetylene LD CH4 Methane est ISO Isoprene LD ETH Ethane LD MACR Methacrolein est* Propane, isobutene, butane, HC3 neopentane, 2,2-dimethylbutane, LD MEK Methyl ethyl ketone LD 2,2-diemthylpentane Isopentane, pentane, propyne, 2-methylpentane, 3-methylpentane, hexane, 2,4-dimethylpentane, HC5 LD MVK Methyl vinyl ketone LD 2,2,3-trimethylbutane, 3,3-dimethylpentane, 2,3-diemthylpentane, isooctane Butyne, cyclopentane + 2,3- diemthylbutane, cyclohexane, α-pinene, β-pinene, HC8 2-methylhexane, heptane, octane, LD API LD 3-carene nonane, undecane, dodecane, nC13, nC14 ETE Ethene LD ROH Borneol LD Propene, 1-butene, isobutene, OLT 3-methyl-1-butene, 1- pentene, LD BENZENE Benzene LD 2- methyl-1-butene, hexene Toluene, chlorobenzene, -

Synthesis of Ethylene Or Propylene/1, 3-Butadiene Copolymers
Catalysts 2015, 5, 2001-2017; doi:10.3390/catal5042001 OPEN ACCESS catalysts ISSN 2073-4344 www.mdpi.com/journal/catalysts Article Synthesis of Ethylene or Propylene/1,3-Butadiene Copolymers Possessing Pendant Vinyl Groups with Virtually No Internal Olefins Kenji Michiue 1,*, Makoto Mitani 2 and Terunori Fujita 2,* 1 Mitsui Chemicals Inc., 1-2, Waki 6-chome, Waki-cho, Kuga-gun, Yamaguchi 740-0061, Japan 2 Mitsui Chemicals Inc., 580-32 Nagaura, Sodegaura, Chiba 299-0265, Japan; E-Mail: [email protected] * Authors to whom correspondence should be addressed; E-Mails: [email protected] (K.M.); [email protected] (T.F.); Tel.: +81-827-53-9121 (K.M.); +81-438-64-2483 (T.F.). Academic Editor: Carl Redshaw Received: 7 October 2015 / Accepted: 12 November 2015 / Published: 20 November 2015 Abstract: In general, ethylene/1,3-butadiene copolymerizations provides copolymers possessing both pendant vinyls and vinylenes as olefinic moieties. We, at MCI, studied the substituent effects of C2-symmetric zirconocene complexes, rac-[Me2Si(Indenyl’)2]ZrCl2 (Indenyl’ = generic substituted indenyl), after activation on the ratio of the pendant vinyls and vinylenes of the resultant copolymers. Complexes examined in this study were rac-dimethylsilylbis (1-indenyl)zirconium dichloride (1), rac-dimethylsilyl-bis[1-(2-methyl-4,5-benzoindenyl)] zirconium dichloride (2), rac-dimethylsilyl-bis[l-(2-methyl -4-phenylindenyl)]zirconium dichloride (3), rac-dimethy1si1y1- bis(2-ethyl-4-phenylindenyl) zirconium dichloride (4), rac-dimethylsilyl-bis[l-(2-n-propyl -4- (1-naphthyl)indenyl)]zirconium dichloride (5), rac-dimethylsilyl-[1-(2-ethyl-4-(5-(2,2- dimethyl-2,3-dihydro-1H-cyclopenta [a]naphthalenyl)indenyl))][1-(2-n-propyl-4-(5-(2,2- dimethyl-2,3-dihydro-1H-cyclopenta[a] naphthalenyl)indenyl))]zirconium dichloride (6), rac-dimethylsilyl-bis[1-(2-ethyl-4-(9-phenanthryl)indenyl)]zirconium dichloride (7), and rac-dimethylsilyl-bis[l-(2-n-propyl-4-(9-phenanthryl)indenyl)]zirconium dichloride (8). -

Cyclopentane Synthesis
Cyclopentane Synthesis Dan O’Malley Baran Group Meeting Cyclopentane Synthesis Group Meeting O'Malley 2/9/2005 This presentation is broken down into the following catagories. Some reactions either fit more than one Students of organic chemistry are taught a number of reactions for the synthesis of category or do not fit easily into any of them. Efforts have been made to place all such reactions in the cyclohexanes at a very early stage of their careers. Techniques for the creation of cyclopentanes, most appropriate category. however, are generally taught at a much later stage and are rarely given the same detailed treatment. This may be the result of the fact that there are no equivalents of reactions such as the Diels-Alder and I. General Information Robinson Annulation in terms of generality, extent of use, and historical importance. This may, in turn, II. Ionic Reactions be caused by the fact that the cyclopentane is an inherintly "umpoled" functionality, as illustrated below. III. Metal Mediated Reactions IV. Radical Reactions FG V. Pericyclic and Pseudo-pericyclic Reactions VI. Ring Expansion and Contraction Reactions I. General Information This situation is further exacerbated by the general lack of cheaply available cyclopentane compounds Baldwin's rules in the chiral pool; wheras a number of cyclohexane terpenes are readily available for elaboration, there Baldwin has divided ring closure reactions into those that are "favored" and those that are "disfavored". are no analogous cylcopentane natural products. Cyclopentanes are however, present in many Those that are disfavored are not always impossible, but are frequently much more difficult to effect.