Organic Chemistry: Air Pollution Studies;
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Allyson M. Buytendyk
DISCOVERING THE ELECTRONIC PROPERTIES OF METAL HYDRIDES, METAL OXIDES AND ORGANIC MOLECULES USING ANION PHOTOELECTRON SPECTROSCOPY by Allyson M. Buytendyk A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland September, 2015 © 2015 Allyson M. Buytendyk All Rights Reserved ABSTRACT Negatively charged molecular ions were studied in the gas phase using anion photoelectron spectroscopy. By coupling theory with the experimentally measured electronic structure, the geometries of the neutral and anion complexes could be predicted. The experiments were conducted using a one-of-a-kind time- of-flight mass spectrometer coupled with a pulsed negative ion photoelectron spectrometer. The molecules studied include metal oxides, metal hydrides, aromatic heterocylic organic compounds, and proton-coupled organic acids. Metal oxides serve as catalysts in reactions from many scientific fields and understanding the catalysis process at the molecular level could help improve reaction efficiencies - - (Chapter 1). The experimental investigation of the super-alkali anions, Li3O and Na3O , revealed both photodetachment and photoionization occur due to the low ionization potential of both neutral molecules. Additionally, HfO- and ZrO- were studied, and although both Hf and Zr have very similar atomic properties, their oxides differ greatly where ZrO- has a much lower electron affinity than HfO-. In the pursuit of using hydrogen as an environmentally friendly fuel alternative, a practical method for storing hydrogen is necessary and metal hydrides are thought to be the answer (Chapter 2). Studies yielding structural and electronic information about the hydrogen - - bonding/interacting in the complex, such as in MgH and AlH4 , are vital to constructing a practical hydrogen storage device. -

Selective Conversion of Acetone to Isobutene and Acetic Acid On
Journal of Catalysis 360 (2018) 66–80 Contents lists available at ScienceDirect Journal of Catalysis journal homepage: www.elsevier.com/locate/jcat Selective conversion of acetone to isobutene and acetic acid on aluminosilicates: Kinetic coupling between acid-catalyzed and radical- mediated pathways ⇑ Stanley Herrmann, Enrique Iglesia Department of Chemical and Biomolecular Engineering, University of California, Berkeley, United States article info abstract Article history: Solid Brønsted acids catalyze aldol condensations that form CAC bonds and remove O-atoms from oxy- Received 1 December 2017 genate reactants, but sequential b-scission reactions also cleave CAC bonds, leading to isobutene and Revised 25 January 2018 acetic acid products for acetone reactants. The elementary steps and site requirements that cause these Accepted 29 January 2018 selectivities to depend sensitively on Al content and framework type in aluminosilicate solid acids remain speculative. Acetone reactions on microporous and mesoporous aluminosilicates (FER, TON, MFI, BEA, MCM-41) showed highest b-scission selectivities on MFI and BEA; they increased as the Al content and Keywords: the intracrystalline density of active protons decreased. The effects of acetone and H O pressure on turn- Solid Brønsted acids 2 over rates and selectivities indicate that an equilibrated pool of reactive C ketols and alkenones are pre- Aldol condensation 6 b-Scission in oxygenates sent at pseudo-steady-state concentrations during catalysis and that they act as intermediates in b- Radical chain cycles scission routes. Two distinct C6 b-scission pathways contribute to the formation of isobutene and acetic Confinement effects acid: (i) a minor H2O-mediated route involving b-scission of C6 ketols on protons and (ii) the predominant anhydrous path, in which H-transfer forms unsaturated C6 enols at protons and these enols propagate radical chains mediated by transition states stabilized by van der Waals contacts within vicinal microp- orous voids. -

Method 323—Measurement of Formaldehyde Emissions from Natural Gas-Fired Stationary Sources—Acetyl Acetone Derivitization Method
While we have taken steps to ensure the accuracy of this Internet version of the document, it is not the official version. Please refer to the official version in the FR publication, which appears on the Government Printing Office's FDSys website (http://www.gpo.gov/fdsys/browse/collectionCfr.action?). Method 323—Measurement of Formaldehyde Emissions From Natural Gas-Fired Stationary Sources—Acetyl Acetone Derivitization Method 1.0 Introduction. This method describes the sampling and analysis procedures of the acetyl acetone colorimetric method for measuring formaldehyde emissions in the exhaust of natural gas-fired, stationary combustion sources. This method, which was prepared by the Gas Research Institute (GRI), is based on the Chilled Impinger Train Method for Methanol, Acetone, Acetaldehyde, Methyl Ethyl Ketone, and Formaldehyde (Technical Bulletin No. 684) developed and published by the National Council of the Paper Industry for Air and Stream Improvement, Inc. (NCASI). However, this method has been prepared specifically for formaldehyde and does not include specifications (e.g., equipment and supplies) and procedures (e.g., sampling and analytical) for methanol, acetone, acetaldehyde, and methyl ethyl ketone. To obtain reliable results, persons using this method should have a thorough knowledge of at least Methods 1 and 2 of 40 CFR part 60, appendix A–1; Method 3 of 40 CFR part 60, appendix A–2; and Method 4 of 40 CFR part 60, appendix A–3. 1.1 Scope and Application 1.1.1 Analytes. The only analyte measured by this method is formaldehyde (CAS Number 50–00–0). 1.1.2 Applicability. This method is for analyzing formaldehyde emissions from uncontrolled and controlled natural gas-fired, stationary combustion sources. -

UNIVERSITY of CALIFORNIA Los Angeles Co-Production of Acetic
UNIVERSITY OF CALIFORNIA Los Angeles Co-production of Acetic Acid and Hydrogen/Power from Natural Gas with Zero Carbon Dioxide Emissions A thesis submitted in partial satisfaction of the requirements for the degree Master of Science in Chemical Engineering by Ibubeleye Somiari 2017 © Copyright by Ibubeleye Somiari 2017 ABSTRACT OF THE THESIS Co-production of Acetic Acid and Hydrogen/Power from Natural Gas with Zero Carbon Dioxide Emissions by Ibubeleye Somiari Master of Science in Chemical Engineering University of California, Los Angeles, 2017 Professor Vasilios Manousiouthakis, Chair In this work, a process plant flow sheet that co-produces acetic acid and hydrogen/power from natural gas with zero carbon dioxide emissions is developed. Two cases are explored: the production of acetic acid and hydrogen (case 1) and the production of acetic acid and power (case 2). This is realized by the selection of an appropriate reaction cluster whose sum results in the overall reaction that co-produces acetic acid and hydrogen/power. The concept of energetic self- sufficiency is introduced and it imposes constraints on the system defined in terms of the ratio of oxygen feed to acetic acid produced. Heat and power integration of the converged flowsheet reveals an operating range for each case that guarantees energetic self-sufficiency. Operating points are chosen to conduct a preliminary economic analysis and a carbon dioxide cost and performance metric calculation to quantify profitability and carbon capture potential of the overall process. ii The thesis of Ibubeleye Somiari is approved. Yvonne Chen Tatiana Segura Vasilios Manousiouthakis, Committee Chair University of California, Los Angeles 2017 iii TABLE OF CONTENTS 1. -

The Decomposition Kinetics of Peracetic Acid and Hydrogen Peroxide in Municipal Wastewaters
Disinfection Forum No 10, October 2015 The Decomposition Kinetics of Peracetic Acid and Hydrogen Peroxide in Municipal Wastewaters INTRODUCTION Efficient control of microbial populations in municipal wastewater using peracetic acid (PAA) requires an understanding of the PAA decomposition kinetics. This knowledge is critical to ensure the proper dosing of PAA needed to achieve an adequate concentration within the contact time of the disinfection chamber. In addition, the impact of PAA on the environment, post-discharge into the receiving water body, also is dependent upon the longevity of the PAA in the environment, before decomposing to acetic acid, oxygen and water. As a result, the decomposition kinetics of PAA may have a significant impact on aquatic and environmental toxicity. PAA is not manufactured as a pure compound. The solution exists as an equilibrium mixture of PAA, hydrogen peroxide, acetic acid, and water: ↔ + + Acetic Acid Hydrogen Peroxide Peracetic Acid Water PeroxyChem’s VigorOx® WWT II Wastewater Disinfection Technology contains 15% peracetic acid by weight and 23% hydrogen peroxide as delivered. Although hydrogen peroxide is present in the formulation, peracetic acid is considered to be the active component for disinfection1 in wastewater. There have been several published studies investigating the decomposition kinetics of PAA in different water matrices, including municipal wastewater2-7. Yuan7 states that PAA may be consumed in the following three competitive reactions: 1. Spontaneous decomposition 2 CH3CO3H à 2 CH3CO2H + O2 Eq (1) 2. Hydrolysis CH3CO3H + H2O à CH3CO2H + H2O2 Eq (2) 3. Transition metal catalyzed decomposition + CH3CO3H + M à CH3CO2H + O2 + other products Eq (3) At neutral pH’s, both peracetic acid and hydrogen peroxide can be rapidly consumed by these reactions7 (hydrogen peroxide will decompose to water and oxygen via 2H2O2 à 2H2O + O2). -

Peracetic Acid Processing
Peracetic Acid Processing Identification Chemical Name(s): CAS Number: peroxyacetic acid, ethaneperoxic acid 79-21-0 Other Names: Other Codes: per acid, periacetic acid, PAA NIOSH Registry Number: SD8750000 TRI Chemical ID: 000079210 UN/ID Number: UN3105 Summary Recommendation Synthetic / Allowed or Suggested Non-Synthetic: Prohibited: Annotation: Synthetic Allowed (consensus) Allowed only for direct food contact for use in wash water. Allowed as a (consensus) sanitizer on surfaces in contact with organic food. (consensus) From hydrogen peroxide and fermented acetic acid sources only. (Not discussed by processing reviewers--see discussion of source under Crops PAA TAP review.) Characterization Composition: C2H4O3. Peracetic acid is a mixture of acetic acid (CH3COOH) and hydrogen peroxide (H2O2) in an aqueous solution. Acetic acid is the principle component of vinegar. Hydrogen peroxide has been previously recommended by the NOSB for the National List in processing (synthetic, allowed at Austin, 1995). Properties: It is a very strong oxidizing agent and has stronger oxidation potential than chlorine or chlorine dioxide. Liquid, clear, and colorless with no foaming capability. It has a strong pungent acetic acid odor, and the pH is acid (2.8). Specific gravity is 1.114 and weighs 9.28 pounds per gallon. Stable upon transport. How Made: Peracetic acid (PAA) is produced by reacting acetic acid and hydrogen peroxide. The reaction is allowed to continue for up to ten days in order to achieve high yields of product according to the following equation. O O || || CH3-C-OH + H2O2 CH3C-O-OH + H2O acetic acid hydrogen peroxyacetic peroxide acid Due to reaction limitations, PAA generation can be up to 15% with residual levels of hydrogen peroxide (up to 25%) and acetic acid (up to 35%) with water up to 25%. -

SAFETY DATA SHEET Cyclopentane
SAFETY DATA SHEET Cyclopentane Section 1. Identification GHS product identifier : Cyclopentane Chemical name : cyclopentane Other means of : cyclopentane (dot); pentamethylene identification Product use : Synthetic/Analytical chemistry. Synonym : cyclopentane (dot); pentamethylene SDS # : 001124 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 2 substance or mixture AQUATIC HAZARD (ACUTE) - Category 3 AQUATIC HAZARD (LONG-TERM) - Category 3 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Highly flammable liquid and vapor. May form explosive mixtures in Air. Harmful to aquatic life with long lasting effects. Precautionary statements General : Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Prevention : Wear protective gloves. Wear eye or face protection. Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. No smoking. Use explosion- proof electrical, ventilating, lighting and all material-handling equipment. Use only non- sparking tools. Take precautionary measures against static discharge. Keep container tightly closed. Avoid release to the environment. Response : IF ON SKIN (or hair): Take off immediately all contaminated clothing. Rinse skin with water or shower. Storage : Store in a well-ventilated place. Keep cool. Disposal : Dispose of contents and container in accordance with all local, regional, national and international regulations. Hazards not otherwise : None known. -
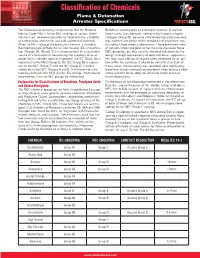
Classification of Chemicals
Classification of Chemicals Flame & Detonation Arrester Specifications PROTECTOSEAL ® The Protectoseal Company recommends that the National Butadiene would qualify as a Group D material. In each of Electric Code (NEC) Article 500, rankings of various chemi - these cases, the chemicals were primarly listed in a higher cals be used, whenever possible, to determine the suitability category (Group B), because of relatively high pressure read - of a detonation arrester for use with a particular chemical. ings noted in one phase of the standard test procedure con - When no NEC rating of the particular chemical is available, ducted by Underwriters Laboratories. These pressures were the International Electrotechnical Commission (IEC) classifica - of concern when categorizing the chemicals because these tion (Groups IIA, IIB and IIC) is recommended as a secondary NEC groupings are also used as standard indicators for the source of information for determining the suitability of an ar - design strength requirements of electrical boxes, apparatus, rester for its intended service. In general, the IEC Group IIA is etc. that must withstand the pressures generated by an igni - equivalent to the NEC Group D; the IEC Group IIB is equiva - tion within the container. It should be noted that, in each of lent to the NEC Group C; and the IEC Group IIC includes these cases, the test pressures recorded were significantly chemicals in the NEC Groups A and B. In the event of a dis - lower than those commonly encountered when testing a deto - crepancy between the NEC and the IEC ratings, Protectoseal nation arrester for its ability to withstand stable and over - recommends that the NEC groups be referenced. -

MSDS Cyclopentane (1).Pdf
MATERIAL SAFETY DATA SHEET (MSDS) CYCLOPENTANE 1. CHEMICAL PRODUCT AND COMPANY IDENTIFICATION SUBSTANCE OR PREPATION TRADE NAME Cyclopentane CHEMICAL CLASSIFICATION Cycloparaffin, Naphthene COMPANY/ UNDERTAKING NAME AND Haldia Petrochemicals Limited, ADDRESS PO Box No 12, Haldia Plant PO Durgachak, Dist Midnapore West Bengal, India PIN 721 602 TELEPHONE 091-3224-274384 / 274400 EMERGENCY TELEPHONE NUMBER 091-3224-275916 2. COMPOSTION AND INFORMATION ON INGREDIENTS CHEMICAL CHEMICLAL CONTENT CAS EXPOSURE LIMITS IN AIR NAME FORMULA NUMBER (ppm) ACGIH ACGIH IDLH TLV- TLV- TWA STEL Cyclopentane C5H10 98.20 wt% 287-92-3 600 NA NA 2,2 Dimethyl (CH3)3CCH2CH3 0.28 wt% 75-83-2 500 1000 NA butane n- pentane n- C5H12 1.47 wt% 109-66-0 600 750 NA i- Pentane i- C5H12 0.05 wt% 78-78-4 600 750 NA Total Sulpher S < 1.0 wt. ppm 7704-34-9 NA NA NA 3. HAZARD CLASSIFICATION Highly flammable liquid and vapour. Vapour EMERGENCY OVERVIEW may cause flash fire. Harmful or Fatal is enter lungs and cause damage POTENTIAL HEALTH HAZARDS EYE SKIN INHALATION INGESTION OTHERS ACUTE To cause Not expected Dizziness, Abdominal prolonged or to be harmful headache, pain, significant to internal nausea, diarrhoea, eye irritation organs if Unconsciousness, dizziness, absorbed weakness nausea, sore through the throat skin. CHRONIC Repeated or prolonged contact with skin may cause dermatitis NFPA HAZARD HEALTH FLAMMABILITY REACTIVITY SPECIAL SIGNALS 2 3 0 - HAZCHEM CODE 3YE GHS-Classification Flammable liquids Category 2 Aspiration hazard Category 1 Chronic aquatic toxicity, Category 3 Acute aquatic toxicity, Category 3 Target Organ Systemic Toxicant - Single exposure, Category 3, Inhalation, Central nervous system Document Compiled By Approved By Issue No Rev. -

Cycloalkanes, Cycloalkenes, and Cycloalkynes
CYCLOALKANES, CYCLOALKENES, AND CYCLOALKYNES any important hydrocarbons, known as cycloalkanes, contain rings of carbon atoms linked together by single bonds. The simple cycloalkanes of formula (CH,), make up a particularly important homologous series in which the chemical properties change in a much more dramatic way with increasing n than do those of the acyclic hydrocarbons CH,(CH,),,-,H. The cyclo- alkanes with small rings (n = 3-6) are of special interest in exhibiting chemical properties intermediate between those of alkanes and alkenes. In this chapter we will show how this behavior can be explained in terms of angle strain and steric hindrance, concepts that have been introduced previously and will be used with increasing frequency as we proceed further. We also discuss the conformations of cycloalkanes, especially cyclo- hexane, in detail because of their importance to the chemistry of many kinds of naturally occurring organic compounds. Some attention also will be paid to polycyclic compounds, substances with more than one ring, and to cyclo- alkenes and cycloalkynes. 12-1 NOMENCLATURE AND PHYSICAL PROPERTIES OF CYCLOALKANES The IUPAC system for naming cycloalkanes and cycloalkenes was presented in some detail in Sections 3-2 and 3-3, and you may wish to review that ma- terial before proceeding further. Additional procedures are required for naming 446 12 Cycloalkanes, Cycloalkenes, and Cycloalkynes Table 12-1 Physical Properties of Alkanes and Cycloalkanes Density, Compounds Bp, "C Mp, "C diO,g ml-' propane cyclopropane butane cyclobutane pentane cyclopentane hexane cyclohexane heptane cycloheptane octane cyclooctane nonane cyclononane "At -40". bUnder pressure. polycyclic compounds, which have rings with common carbons, and these will be discussed later in this chapter. -

Cyclopentane Cyp
CYCLOPENTANE CYP CAUTIONARY RESPONSE INFORMATION 4. FIRE HAZARDS 7. SHIPPING INFORMATION 4.1 Flash Point: 7.1 Grades of Purity: Commercial; 60% (remainder Common Synonyms Watery liquid Colorless Mild, sweet odor < 20°F C.C. consists of hydrocarbons of similar boiling Pentamethylene 4.2 Flammable Limits in Air: (approx.) 1.1%- point); Research: 99+% 8.7% 7.2 Storage Temperature: Ambient Floats on water. Flammable, irritating vapor is produced. 4.3 Fire Extinguishing Agents: Dry 7.3 Inert Atmosphere: No requirement chemical, foam, carbon dioxide Keep people away. 7.4 Venting: Pressure-vacuum Avoid inhalation. 4.4 Fire Extinguishing Agents Not to Be Shut off ingition sources. Call fire department. Used: Water may be ineffective. 7.5 IMO Pollution Category: (C) Evacuate area in case of large discharge. 4.5 Special Hazards of Combustion 7.6 Ship Type: 3 Notify local health and pollution control agencies. Products: Not pertinent 7.7 Barge Hull Type: Currently not available Protect water intakes. 4.6 Behavior in Fire: Containers may explode. FLAMMABLE. 8. HAZARD CLASSIFICATIONS Fire 4.7 Auto Ignition Temperature: 682°F Containers may explode in fire. 8.1 49 CFR Category: Flammable liquid Flashback along vapor trail may occur. 4.8 Electrical Hazards: Not pertinent 8.2 49 CFR Class: 3 Vapor may explode if ignited in an enclosed area. 4.9 Burning Rate: 7.9 mm/min. 8.3 49 CFR Package Group: II Extinguish with dry chemicals, foam or carbon dioxide. 4.10 Adiabatic Flame Temperature: Currently Water may be ineffective on fire. not available 8.4 Marine Pollutant: No Cool exposed containers with water. -

Section 2. Hazards Identification OSHA/HCS Status : This Material Is Considered Hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200)
SAFETY DATA SHEET Flammable Liquefied Gas Mixture: 2-Methylpentane / 2-Methylhexane / 2, 3-Dimethylheptane / Benzene / Cyclohexane / Cyclopentane / Dodecane / Ethyl Benzene / Ethyl Cyclohexane / Hexane / Isobutane / Isobutyl Cyclohexane / Isooctane / Isopentane / Isopropylcyclohexane / Methyl Cyclohexane / N-Butane / N-Butyl Benzene / N-Decane / N-Heptane / N-Nonane / N-Octane / N-Pentane / N-Propyl Benzene / Toluene Section 1. Identification GHS product identifier : Flammable Liquefied Gas Mixture: 2-Methylpentane / 2-Methylhexane / 2, 3-Dimethylheptane / Benzene / Cyclohexane / Cyclopentane / Dodecane / Ethyl Benzene / Ethyl Cyclohexane / Hexane / Isobutane / Isobutyl Cyclohexane / Isooctane / Isopentane / Isopropylcyclohexane / Methyl Cyclohexane / N-Butane / N-Butyl Benzene / N-Decane / N-Heptane / N-Nonane / N-Octane / N-Pentane / N-Propyl Benzene / Toluene Other means of : Not available. identification Product type : Liquefied gas Product use : Synthetic/Analytical chemistry. SDS # : 021986 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE GASES - Category 1 substance or mixture GASES UNDER PRESSURE - Liquefied gas SKIN IRRITATION - Category 2 GERM CELL MUTAGENICITY - Category 1 CARCINOGENICITY - Category 1 TOXIC TO REPRODUCTION (Fertility) - Category 2 TOXIC TO REPRODUCTION (Unborn child) - Category 2 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (REPEATED EXPOSURE) - Category 1 AQUATIC HAZARD (ACUTE) - Category 2 AQUATIC HAZARD (LONG-TERM) - Category 2 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable gas. May form explosive mixtures with air.