Method 515.4 Determination of Chlorinated Acids In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

2,4-Dichlorophenoxyacetic Acid
2,4-Dichlorophenoxyacetic acid 2,4-Dichlorophenoxyacetic acid IUPAC (2,4-dichlorophenoxy)acetic acid name 2,4-D Other hedonal names trinoxol Identifiers CAS [94-75-7] number SMILES OC(COC1=CC=C(Cl)C=C1Cl)=O ChemSpider 1441 ID Properties Molecular C H Cl O formula 8 6 2 3 Molar mass 221.04 g mol−1 Appearance white to yellow powder Melting point 140.5 °C (413.5 K) Boiling 160 °C (0.4 mm Hg) point Solubility in 900 mg/L (25 °C) water Related compounds Related 2,4,5-T, Dichlorprop compounds Except where noted otherwise, data are given for materials in their standard state (at 25 °C, 100 kPa) 2,4-Dichlorophenoxyacetic acid (2,4-D) is a common systemic herbicide used in the control of broadleaf weeds. It is the most widely used herbicide in the world, and the third most commonly used in North America.[1] 2,4-D is also an important synthetic auxin, often used in laboratories for plant research and as a supplement in plant cell culture media such as MS medium. History 2,4-D was developed during World War II by a British team at Rothamsted Experimental Station, under the leadership of Judah Hirsch Quastel, aiming to increase crop yields for a nation at war.[citation needed] When it was commercially released in 1946, it became the first successful selective herbicide and allowed for greatly enhanced weed control in wheat, maize (corn), rice, and similar cereal grass crop, because it only kills dicots, leaving behind monocots. Mechanism of herbicide action 2,4-D is a synthetic auxin, which is a class of plant growth regulators. -

Common and Chemical Names of Herbicides Approved by the WSSA
Weed Science 2010 58:511–518 Common and Chemical Names of Herbicides Approved by the Weed Science Society of America Below is the complete list of all common and chemical of herbicides as approved by the International Organization names of herbicides approved by the Weed Science Society of for Standardization (ISO). A sponsor may submit a proposal America (WSSA) and updated as of September 1, 2010. for a common name directly to the WSSA Terminology Beginning in 1996, it has been published yearly in the last Committee. issue of Weed Science with Directions for Contributors to A herbicide common name is not synonymous with Weed Science. This list is published in lieu of the selections a commercial formulation of the same herbicide, and in printed previously on the back cover of Weed Science. Only many instances, is not synonymous with the active ingredient common and chemical names included in this complete of a commercial formulation as identified on the product list should be used in WSSA publications. In the absence of label. If the herbicide is a salt or simple ester of a parent a WSSA-approved common name, the industry code number compound, the WSSA common name applies to the parent as compiled by the Chemical Abstracts Service (CAS) with compound only. CAS systematic chemical name or the systematic chemical The chemical name used in this list is that preferred by the name alone may be used. The current approved list is also Chemical Abstracts Service (CAS) according to their system of available at our web site (www.wssa.net). -

Herbicide Mode of Action Table High Resistance Risk
Herbicide Mode of Action Table High resistance risk Chemical family Active constituent (first registered trade name) GROUP 1 Inhibition of acetyl co-enzyme A carboxylase (ACC’ase inhibitors) clodinafop (Topik®), cyhalofop (Agixa®*, Barnstorm®), diclofop (Cheetah® Gold* Decision®*, Hoegrass®), Aryloxyphenoxy- fenoxaprop (Cheetah®, Gold*, Wildcat®), fluazifop propionates (FOPs) (Fusilade®), haloxyfop (Verdict®), propaquizafop (Shogun®), quizalofop (Targa®) Cyclohexanediones (DIMs) butroxydim (Factor®*), clethodim (Select®), profoxydim (Aura®), sethoxydim (Cheetah® Gold*, Decision®*), tralkoxydim (Achieve®) Phenylpyrazoles (DENs) pinoxaden (Axial®) GROUP 2 Inhibition of acetolactate synthase (ALS inhibitors), acetohydroxyacid synthase (AHAS) Imidazolinones (IMIs) imazamox (Intervix®*, Raptor®), imazapic (Bobcat I-Maxx®*, Flame®, Midas®*, OnDuty®*), imazapyr (Arsenal Xpress®*, Intervix®*, Lightning®*, Midas®* OnDuty®*), imazethapyr (Lightning®*, Spinnaker®) Pyrimidinyl–thio- bispyribac (Nominee®), pyrithiobac (Staple®) benzoates Sulfonylureas (SUs) azimsulfuron (Gulliver®), bensulfuron (Londax®), chlorsulfuron (Glean®), ethoxysulfuron (Hero®), foramsulfuron (Tribute®), halosulfuron (Sempra®), iodosulfuron (Hussar®), mesosulfuron (Atlantis®), metsulfuron (Ally®, Harmony®* M, Stinger®*, Trounce®*, Ultimate Brushweed®* Herbicide), prosulfuron (Casper®*), rimsulfuron (Titus®), sulfometuron (Oust®, Eucmix Pre Plant®*, Trimac Plus®*), sulfosulfuron (Monza®), thifensulfuron (Harmony®* M), triasulfuron (Logran®, Logran® B-Power®*), tribenuron (Express®), -

Agricultural Biotechnology: Benefits of Transgenic Soybeans
AGRICULTURAL BIOTECHNOLOGY: BENEFITS OF TRANSGENIC SOYBEANS Leonard P. Gianessi Janet E. Carpenter April 2000 National Center for Food and Agricultural Policy 1616 P Street, NW, First Floor Washington, DC 20036 Tel: 202-328-5048 Fax: 202-328-5133 [email protected] Preparation of this report was supported financially with a grant from the Rockefeller Foundation TABLE OF CONTENTS 1. Introduction 2. U.S. Soybean Production 3. Soybean Products 4. Soybean Physiology 5. Soybeans – Agronomic Factors 6. Soybean Genetic Improvements A. Introduction B. Reproductive Process C. Artificial Cross Breeding D. Mutation Breeding E. Transgenic Plants 7. Weed Competition – Soybeans 8. Weed Control in Soybeans: 1940’s – 1950’s 9. Herbicides – An Overview 10. Herbicide Use in Soybeans: 1960’s – 1995 A. Introduction B. Historical Overview 1. The Early 1960’s 2. Soil Applied Herbicides 3. Postemergence Herbicides 4. Sulfonylurea/Imidazolinone Herbicides 5. Burndown Herbicides C. Summary of Usage: 1995 11. Transgenic Herbicide Tolerant Soybeans A. Glyphosate – An Overview B. Performance of Roundup Ready Soybeans C. Herbicide Ratings D. Adoption Impacts: 1995 – 1998 1. Herbicide Costs 2. Soybean Yields 3. Returns 4. Other Aggregate Studies 5. Herbicide Treatments 6. Herbicide Use Amounts 7. Other Impacts 12. Summary and Conclusions 13. References Appendix 1: Soybean Processing – A Description 1. Introduction Soybeans and other crops have been improved genetically for many decades through traditional crop breeding – a technique that requires that species be sexually compatible. With the development of biotechnology methods, scientists have the ability to transfer single genes from one living organism into another, regardless of species or sexual compatibility. Varieties that are developed through the transfer of genes between species that are not sexually compatible are referred to as “transgenic.” Transgenic soybean plants have been developed with a gene from a soil bacteria that allows the use of an herbicide that would normally kill soybeans. -

INDEX to PESTICIDE TYPES and FAMILIES and PART 180 TOLERANCE INFORMATION of PESTICIDE CHEMICALS in FOOD and FEED COMMODITIES
US Environmental Protection Agency Office of Pesticide Programs INDEX to PESTICIDE TYPES and FAMILIES and PART 180 TOLERANCE INFORMATION of PESTICIDE CHEMICALS in FOOD and FEED COMMODITIES Note: Pesticide tolerance information is updated in the Code of Federal Regulations on a weekly basis. EPA plans to update these indexes biannually. These indexes are current as of the date indicated in the pdf file. For the latest information on pesticide tolerances, please check the electronic Code of Federal Regulations (eCFR) at http://www.access.gpo.gov/nara/cfr/waisidx_07/40cfrv23_07.html 1 40 CFR Type Family Common name CAS Number PC code 180.163 Acaricide bridged diphenyl Dicofol (1,1-Bis(chlorophenyl)-2,2,2-trichloroethanol) 115-32-2 10501 180.198 Acaricide phosphonate Trichlorfon 52-68-6 57901 180.259 Acaricide sulfite ester Propargite 2312-35-8 97601 180.446 Acaricide tetrazine Clofentezine 74115-24-5 125501 180.448 Acaricide thiazolidine Hexythiazox 78587-05-0 128849 180.517 Acaricide phenylpyrazole Fipronil 120068-37-3 129121 180.566 Acaricide pyrazole Fenpyroximate 134098-61-6 129131 180.572 Acaricide carbazate Bifenazate 149877-41-8 586 180.593 Acaricide unclassified Etoxazole 153233-91-1 107091 180.599 Acaricide unclassified Acequinocyl 57960-19-7 6329 180.341 Acaricide, fungicide dinitrophenol Dinocap (2, 4-Dinitro-6-octylphenyl crotonate and 2,6-dinitro-4- 39300-45-3 36001 octylphenyl crotonate} 180.111 Acaricide, insecticide organophosphorus Malathion 121-75-5 57701 180.182 Acaricide, insecticide cyclodiene Endosulfan 115-29-7 79401 -
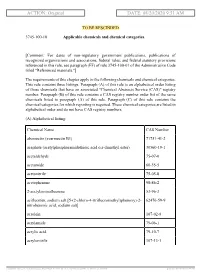
ACTION: Original DATE: 08/20/2020 9:51 AM
ACTION: Original DATE: 08/20/2020 9:51 AM TO BE RESCINDED 3745-100-10 Applicable chemicals and chemical categories. [Comment: For dates of non-regulatory government publications, publications of recognized organizations and associations, federal rules, and federal statutory provisions referenced in this rule, see paragraph (FF) of rule 3745-100-01 of the Administrative Code titled "Referenced materials."] The requirements of this chapter apply to the following chemicals and chemical categories. This rule contains three listings. Paragraph (A) of this rule is an alphabetical order listing of those chemicals that have an associated "Chemical Abstracts Service (CAS)" registry number. Paragraph (B) of this rule contains a CAS registry number order list of the same chemicals listed in paragraph (A) of this rule. Paragraph (C) of this rule contains the chemical categories for which reporting is required. These chemical categories are listed in alphabetical order and do not have CAS registry numbers. (A) Alphabetical listing: Chemical Name CAS Number abamectin (avermectin B1) 71751-41-2 acephate (acetylphosphoramidothioic acid o,s-dimethyl ester) 30560-19-1 acetaldehyde 75-07-0 acetamide 60-35-5 acetonitrile 75-05-8 acetophenone 98-86-2 2-acetylaminofluorene 53-96-3 acifluorfen, sodium salt [5-(2-chloro-4-(trifluoromethyl)phenoxy)-2- 62476-59-9 nitrobenzoic acid, sodium salt] acrolein 107-02-8 acrylamide 79-06-1 acrylic acid 79-10-7 acrylonitrile 107-13-1 [ stylesheet: rule.xsl 2.14, authoring tool: RAS XMetaL R2_0F1, (dv: 0, p: 185720, pa: -

U.S. EPA, Pesticide Product Label, QUINCLORAC 75 SWF, 02/06/2006
u.s. ENVIRONM£~J7AL PkO';EC';ION N:JENCY EPfI, Req. Cate ~f Issuance: Office of t'esticide "roqram.<; Number: Keqistratlon Divlsion (7:'()SC) 1200 Pennsylvania Ave., N.W. WashingtrJII, D.::'. ?046D 42750- 131 FEE - 6 2006 NOTICE OF PESTICIDE: Term of Issuance: ~ Registration Conditional __ Reregistration : under r: fRJ'l., as am'?nd'O'd: Name of ~esticide Product: Quinclorac 75DF SWF Name and Address of RegIstrant ,include ZIP Codej: Albaugh, Inc. P.O. Box 2127 Valdosta, GA 31604-2127 Note: Changes in .LabEoli~fg differing in suostance from that accepted in connection with this registration must be submitted to and accepted by the Registratlon Division prior to use of the label in commerce. In any correspondence on this product always refer to the above EPA registration number. em ::he bas':s of lnformat':on f ~rr,lshed by the reg:s'Crant, the above llamed pesticide ':5 hereby roegisterea/reregJ.3terec under :::--.e federa; =nsec::':c:ide, fungicide and Rodenticide Act. Reg':straL,on is ir. no (.Jay tc be ::onstrued as an endorsement or recommendation of this product by the Agency. =n urder to protect hea~ttJ and tr.e ",nvil"onment, ::he Admir.istrator, on his motion, may at any time suspend or cancel the regi3t:::ation of a pesticide in accoraance with the Act. The acceptance of any name 1.n connection with the registration of a product under this Act is not to be construed as giving the registrant a right to -=xclusive use of the r,ame or to its use if it has been coveroed by others. -

US EPA, Pesticide Product Label, QUINCLORAC 4L AG,03/04/2019
81,7('67$7(6(19,5210(17$/3527(&7,21$*(1&< :$6+,1*721'& 2)),&(2)&+(0,&$/6$)(7< $1'32//87,2135(9(17,21 0DUFK 0RUULV*DVNLQV $OEDXJK//& 32%R[ 9DOGRVWD*$ 6XEMHFW /DEHO$PHQGPHQW±5HLQVWDWH3DVWXUHDQG5DQJHODQG8VH 3URGXFW1DPH48,1&/25$&/$* (3$5HJLVWUDWLRQ1XPEHU $SSOLFDWLRQ'DWH 'HFLVLRQ1XPEHU 'HDU0U*DVNLQV 7KHDPHQGHGODEHOUHIHUUHGWRDERYHVXEPLWWHGLQFRQQHFWLRQZLWKUHJLVWUDWLRQXQGHUWKH)HGHUDO ,QVHFWLFLGH)XQJLFLGHDQG5RGHQWLFLGH$FWDVDPHQGHGLVDFFHSWDEOH7KLVDSSURYDOGRHVQRW DIIHFWDQ\FRQGLWLRQVWKDWZHUHSUHYLRXVO\LPSRVHGRQWKLVUHJLVWUDWLRQ<RXFRQWLQXHWREH VXEMHFWWRH[LVWLQJFRQGLWLRQVRQ\RXUUHJLVWUDWLRQDQGDQ\GHDGOLQHVFRQQHFWHGZLWKWKHP $VWDPSHGFRS\RI\RXUODEHOLQJLVHQFORVHGIRU\RXUUHFRUGV7KLVODEHOLQJVXSHUVHGHVDOO SUHYLRXVO\DFFHSWHGODEHOLQJ<RXPXVWVXEPLWRQHFRS\RIWKHILQDOSULQWHGODEHOLQJEHIRUH\RX UHOHDVHWKHSURGXFWIRUVKLSPHQWZLWKWKHQHZODEHOLQJ,QDFFRUGDQFHZLWK&)5 F \RXPD\GLVWULEXWHRUVHOOWKLVSURGXFWXQGHUWKHSUHYLRXVO\DSSURYHGODEHOLQJIRUPRQWKV IURPWKHGDWHRIWKLVOHWWHU$IWHUPRQWKV\RXPD\RQO\GLVWULEXWHRUVHOOWKLVSURGXFWLILW EHDUVWKLVQHZUHYLVHGODEHOLQJRUVXEVHTXHQWO\DSSURYHGODEHOLQJ³7RGLVWULEXWHRUVHOO´LV GHILQHGXQGHU),)5$VHFWLRQ JJ DQGLWVLPSOHPHQWLQJUHJXODWLRQDW&)5 6KRXOG\RXZLVKWRDGGUHWDLQDUHIHUHQFHWRWKHFRPSDQ\¶VZHEVLWHRQ\RXUODEHOWKHQSOHDVHEH DZDUHWKDWWKHZHEVLWHEHFRPHVODEHOLQJXQGHUWKH)HGHUDO,QVHFWLFLGH)XQJLFLGHDQG5RGHQWLFLGH $FWDQGLVVXEMHFWWRUHYLHZE\WKH$JHQF\,IWKHZHEVLWHLVIDOVHRUPLVOHDGLQJWKHSURGXFW ZRXOGEHPLVEUDQGHGDQGXQODZIXOWRVHOORUGLVWULEXWHXQGHU),)5$VHFWLRQ D ( &)5 D OLVWH[DPSOHVRIVWDWHPHQWV(3$PD\FRQVLGHUIDOVHRUPLVOHDGLQJ,QDGGLWLRQ UHJDUGOHVVRIZKHWKHUDZHEVLWHLVUHIHUHQFHGRQ\RXUSURGXFW¶VODEHOFODLPVPDGHRQWKH -

Thickening Glyphosate Formulations
(19) TZZ _T (11) EP 2 959 777 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 30.12.2015 Bulletin 2015/53 A01N 57/20 (2006.01) A01N 25/30 (2006.01) A01P 13/00 (2006.01) (21) Application number: 15175726.7 (22) Date of filing: 17.08.2009 (84) Designated Contracting States: (71) Applicant: Akzo Nobel N.V. AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 6824 BM Arnhem (NL) HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO SE SI SK SM TR (72) Inventor: ZHU, Shawn Stormville, NY New York 12582 (US) (30) Priority: 19.08.2008 US 90010 P 09.09.2008 EP 08163910 (74) Representative: Akzo Nobel IP Department Velperweg 76 (62) Document number(s) of the earlier application(s) in 6824 BM Arnhem (NL) accordance with Art. 76 EPC: 11191518.7 / 2 425 716 Remarks: 09781884.3 / 2 315 524 This application was filed on 07-07-2015 as a divisional application to the application mentioned under INID code 62. (54) THICKENING GLYPHOSATE FORMULATIONS (57) The present invention generally relates to a glyphosate formulation with enhanced viscosity, said formulation containing a thickening composition comprising at least one nitrogen- containing surfactant. EP 2 959 777 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 959 777 A1 Description FIELD OF THE INVENTION 5 [0001] The present invention relates to a glyphosate formulations thickened by nitrogen containing surfactants. BACKGROUND OF THE INVENTION [0002] Glyphosate is the most widely used herbicide in the world. -

AP-42, CH 9.2.2: Pesticide Application
9.2.2PesticideApplication 9.2.2.1General1-2 Pesticidesaresubstancesormixturesusedtocontrolplantandanimallifeforthepurposesof increasingandimprovingagriculturalproduction,protectingpublichealthfrompest-bornediseaseand discomfort,reducingpropertydamagecausedbypests,andimprovingtheaestheticqualityofoutdoor orindoorsurroundings.Pesticidesareusedwidelyinagriculture,byhomeowners,byindustry,andby governmentagencies.Thelargestusageofchemicalswithpesticidalactivity,byweightof"active ingredient"(AI),isinagriculture.Agriculturalpesticidesareusedforcost-effectivecontrolofweeds, insects,mites,fungi,nematodes,andotherthreatstotheyield,quality,orsafetyoffood.Theannual U.S.usageofpesticideAIs(i.e.,insecticides,herbicides,andfungicides)isover800millionpounds. AiremissionsfrompesticideusearisebecauseofthevolatilenatureofmanyAIs,solvents, andotheradditivesusedinformulations,andofthedustynatureofsomeformulations.Mostmodern pesticidesareorganiccompounds.EmissionscanresultdirectlyduringapplicationorastheAIor solventvolatilizesovertimefromsoilandvegetation.Thisdiscussionwillfocusonemissionfactors forvolatilization.Thereareinsufficientdataavailableonparticulateemissionstopermitemission factordevelopment. 9.2.2.2ProcessDescription3-6 ApplicationMethods- Pesticideapplicationmethodsvaryaccordingtothetargetpestandtothecroporothervalue tobeprotected.Insomecases,thepesticideisapplieddirectlytothepest,andinotherstothehost plant.Instillothers,itisusedonthesoilorinanenclosedairspace.Pesticidemanufacturershave developedvariousformulationsofAIstomeetboththepestcontrolneedsandthepreferred -

List of Herbicide Groups
List of herbicides Group Scientific name Trade name clodinafop (Topik®), cyhalofop (Barnstorm®), diclofop (Cheetah® Gold*, Decision®*, Hoegrass®), fenoxaprop (Cheetah® Gold* , Wildcat®), A Aryloxyphenoxypropionates fluazifop (Fusilade®, Fusion®*), haloxyfop (Verdict®), propaquizafop (Shogun®), quizalofop (Targa®) butroxydim (Falcon®, Fusion®*), clethodim (Select®), profoxydim A Cyclohexanediones (Aura®), sethoxydim (Cheetah® Gold*, Decision®*), tralkoxydim (Achieve®) A Phenylpyrazoles pinoxaden (Axial®) azimsulfuron (Gulliver®), bensulfuron (Londax®), chlorsulfuron (Glean®), ethoxysulfuron (Hero®), foramsulfuron (Tribute®), halosulfuron (Sempra®), iodosulfuron (Hussar®), mesosulfuron (Atlantis®), metsulfuron (Ally®, Harmony®* M, Stinger®*, Trounce®*, B Sulfonylureas Ultimate Brushweed®* Herbicide), prosulfuron (Casper®*), rimsulfuron (Titus®), sulfometuron (Oust®, Eucmix Pre Plant®*), sulfosulfuron (Monza®), thifensulfuron (Harmony®* M), triasulfuron, (Logran®, Logran® B Power®*), tribenuron (Express®), trifloxysulfuron (Envoke®, Krismat®*) florasulam (Paradigm®*, Vortex®*, X-Pand®*), flumetsulam B Triazolopyrimidines (Broadstrike®), metosulam (Eclipse®), pyroxsulam (Crusader®Rexade®*) imazamox (Intervix®*, Raptor®,), imazapic (Bobcat I-Maxx®*, Flame®, Midas®*, OnDuty®*), imazapyr (Arsenal Xpress®*, Intervix®*, B Imidazolinones Lightning®*, Midas®*, OnDuty®*), imazethapyr (Lightning®*, Spinnaker®) B Pyrimidinylthiobenzoates bispyribac (Nominee®), pyrithiobac (Staple®) C Amides: propanil (Stam®) C Benzothiadiazinones: bentazone (Basagran®, -

Pesticides in Iowa's Water Resources: Historical Trends and New Issues
PesticidesPesticides inin IowaIowa’’ss WaterWater Resources:Resources: HistoricalHistorical TrendsTrends andand NewNew IssuesIssues MaryMary P.P. SkopecSkopec andand KatieKatie ForemanForeman WatershedWatershed MonitoringMonitoring andand AssessmentAssessment IowaIowa DepartmentDepartment ofof NaturalNatural ResourcesResources OutlineOutline PesticidePesticide UseUse inin IowaIowa CurrentCurrent StatusStatus andand HistoricalHistorical TrendsTrends NewNew ApproachesApproaches forfor UnderstandingUnderstanding Occurrence of Pesticides in Water Resources Function of: 1. Use 2. Soil Characteristics 3. Rainfall Timing and Amount 4. Pesticide Properties Land Use in Iowa (2002) Percent of Iowa Acres Treated Herbicides Insecticides Yes No Source: Pesticide Movement: What Farmers Need to Know, Iowa State Extension, PAT-36 2000, Non-City Stream Sites 2001, Non-City Stream Sites 2002, Non-City Stream Sites 2,4-D* 2,4,5-T* Acetochlor Acifluoren* Alachlor Ametryn Atrazine Atrazine, Deethyl Atrazine, Deisopropyl Bentazon* Bromacil* Some Bromoxynil* Butylate Very Chloramben* Cyanazine Dalapon* Common; Herbicides Dicamba* Dichloprop* Others Dimethenamid EPTC Less so Metolachlor Metribuzin Pendimethalin* Picloram* Prometon Propachlor Propazine Simazine Triclopyr* Trifluralin Carbaryl* Carbofuran* Chlordane* Chlorpyrifos* es DDD* DDE* id DDT* Few ic Diazinon* ct Dieldrin* Detects Fonofos* Lindane (gamma-BHC)* Inse Malathion* Parathion* Phorate* 0 204060801000 204060801000 20406080100 Detection Rate, in Percent Detection Rate, in Percent Detection Rate,