Genome-Wide Functional Analysis of Human Cell-Cycle Regulators
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analyses of Allele-Specific Gene Expression in Highly Divergent
ARTICLES Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance James J Crowley1,10, Vasyl Zhabotynsky1,10, Wei Sun1,2,10, Shunping Huang3, Isa Kemal Pakatci3, Yunjung Kim1, Jeremy R Wang3, Andrew P Morgan1,4,5, John D Calaway1,4,5, David L Aylor1,9, Zaining Yun1, Timothy A Bell1,4,5, Ryan J Buus1,4,5, Mark E Calaway1,4,5, John P Didion1,4,5, Terry J Gooch1,4,5, Stephanie D Hansen1,4,5, Nashiya N Robinson1,4,5, Ginger D Shaw1,4,5, Jason S Spence1, Corey R Quackenbush1, Cordelia J Barrick1, Randal J Nonneman1, Kyungsu Kim2, James Xenakis2, Yuying Xie1, William Valdar1,4, Alan B Lenarcic1, Wei Wang3,9, Catherine E Welsh3, Chen-Ping Fu3, Zhaojun Zhang3, James Holt3, Zhishan Guo3, David W Threadgill6, Lisa M Tarantino7, Darla R Miller1,4,5, Fei Zou2,11, Leonard McMillan3,11, Patrick F Sullivan1,5,7,8,11 & Fernando Pardo-Manuel de Villena1,4,5,11 Complex human traits are influenced by variation in regulatory DNA through mechanisms that are not fully understood. Because regulatory elements are conserved between humans and mice, a thorough annotation of cis regulatory variants in mice could aid in further characterizing these mechanisms. Here we provide a detailed portrait of mouse gene expression across multiple tissues in a three-way diallel. Greater than 80% of mouse genes have cis regulatory variation. Effects from these variants influence complex traits and usually extend to the human ortholog. Further, we estimate that at least one in every thousand SNPs creates a cis regulatory effect. -

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

By Submitted in Partial Satisfaction of the Requirements for Degree of in In
Developments of Two Imaging based Technologies for Cell Biology Researches by Xiaowei Yan DISSERTATION Submitted in partial satisfaction of the requirements for degree of DOCTOR OF PHILOSOPHY in Biochemistry and Molecular Biology in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________Ronald Vale Chair ______________________________________________________________________________Jonathan Weissman ______________________________________________________________________________Orion Weiner ______________________________________________________________________________ ______________________________________________________________________________ Committee Members Copyright 2021 By Xiaowei Yan ii DEDICATION Everything happens for the best. To my family, who supported me with all their love. iii ACKNOWLEDGEMENTS The greatest joy of my PhD has been joining UCSF, working and learning with such a fantastic group of scientists. I am extremely grateful for all the support and mentorship I received and would like to thank: My mentor, Ron Vale, who is such a great and generous person. Thank you for showing me that science is so much fun and thank you for always giving me the freedom in pursuing my interest. I am grateful for all the guidance from you and thank you for always supporting me whenever I needed. You are a person full of wisdom, and I have been learning so much from you and your attitude to science, science community and even life will continue inspire me. Thank you for being my mentor and thank you for being such a great mentor. Everyone else in Vale lab, past and present, for making our lab a sweet home. I would like to give my special thank to Marvin (Marvin Tanenbaum) and Nico (Nico Stuurman), two other mentors for me in the lab. I would like to thank them for helping me adapt to our lab, for all the valuable advice and for all the happiness during the time that we work together. -
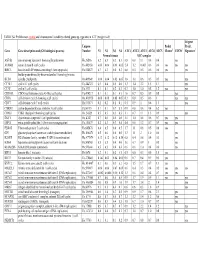
TABLE S4: Proliferation-Related and Chromosomal Instability-Related
TABLE S4: Proliferation-related and chromosomal instability-related genes up-regulated in ATC (weight <4.0) 44-gene Unigene Prolif. Prolif. Gene Gene description and (GO biological process) Number N1 N2 N3 N4 ATC1 ATC2 ATC3 ATC4 ATC5 Cluster1 CIN702 Signature3 Normal tissues ATC samples ASF1B anti-silencing function 1 homolog B (unknown) Hs.26516 0.2 0.3 0.3 0.3 0.8 0.8 1.1 0.8 0.4 yes AURKB aurora kinase B (cell cycle) Hs.442658 0.06 0.06 0.08 0.05 2.4 1.2 0.003 0.6 0.6 yes yes yes BIRC5 baculoviral IAP repeat-containing 5 (anti apoptosis) Hs.514527 0.7 0.3 0.4 0.3 0.8 0.5 0.5 0.6 0.6 yes yes budding uninhibited by benzimidazoles 1 homolog (mitotic BUB1 spindle checkpoint) Hs.469649 0.06 0.04 0.02 0.05 1.8 1.0 0.6 0.5 0.5 yes yes CCNE1 cyclin E1 (cell cycle) Hs.244723 0.5 0.4 0.5 0.6 1.3 1.4 2.2 1.5 1.1 yes CCNF cyclin F (cell cycle) Hs.1973 0.1 0.1 0.2 0.3 0.7 1.0 1.0 0.8 1.2 yes yes CDC45L CDC45 cell division cycle 45-like (cell cycle) Hs.474217 0.1 0.1 0.1 0.3 1.6 0.7 0.5 0.9 0.8 yes CDC6 cell division cycle 6 homolog (cell cycle) Hs. 405958 0.08 0.08 0.08 0.07 0.3 0.8 0.5 0.6 1 yes yes CDC7 cell division cycle 7 (cell cycle) Hs.533573 0.2 0.2 0.2 0.1 0.5 0.9 1 0.6 1.3 yes CDKN3 cyclin-dependent kinase inhibitor 3 (cell cycle) Hs.84113 0.1 0.1 0.7 0.1 0.9 0.8 0.6 0.4 6.2 yes CHEK1 CHK1 checkpoint homolog (cell cycle) Hs. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -
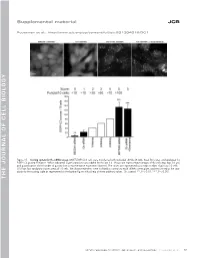
Thejournalofcellb Io Logy
Supplemental material JCB Puustinen et al., http://www.jcb.org/cgi/content/full/jcb.201304012/DC1 Figure S1. Scoring system for the siRNA screen. MCF7-EGFP-LC3 cells were transfected with indicated siRNAs (8 nM), fixed 56 h later, and analyzed for EGFP-LC3 puncta formation. When indicated, 5 µM siramesine was added for the last 3 h. Shown are representative images of the cells (top; bar, 10 µm) and quantification of the number of puncta from a representative experiment (bottom). The values are represented as a mean number of puncta/10 cells ± SDs from four randomly chosen areas of 10 cells. The shown treatments were included as controls to each siRNA screen plate, and they served as the stan- dards for the scoring scale as represented on the bottom figure with a help of three arbitrary values. Ctr, control. **, P < 0.01; ***, P < 0.001. THE JOURNAL OF CELL BIOLOGY CIP2A regulates mTORC1, cell growth, and autophagy • Puustinen et al. S1 Figure S2. A cartoon highlighting the positions of the candidate autophagy-regulating genes in the insulin receptor signaling network. The cartoon was prepared by Ingenuity Pathway Analysis. software. CP, carbohydrate phosphatase; ERK, extracellular signal–regulated kinase; FSH, follicle-stimulating hor- mone; EG, epidermal growth factor. S2 JCB Figure S3. The effect of siRNAs targeting the autophagy-regulating PP2 subunits on target gene expression, cell density, and mTORC1 pathway. (A) Total RNA was isolated from MCF7 cells treated with indicated siRNAs (20 nM) for 54 h and analyzed for target gene expression by qPCR using primers de- signed to specifically amplify the indicated PP2A-related mRNAs andACTB mRNA (internal control). -

Comparative Analysis of Human Chromosome 7Q21 and Mouse
Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Comparative analysis of human chromosome 7q21 and mouse proximal chromosome 6 reveals a placental-specific imprinted gene, TFPI2/Tfpi2, which requires EHMT2 and EED for allelic-silencing David Monk,1,6 Alexandre Wagschal,2 Philippe Arnaud,2 Pari-Sima Mu¨ller,3 Layla Parker-Katiraee,4 Déborah Bourc’his,5 Stephen W. Scherer,4 Robert Feil,2 Philip Stanier,1 and Gudrun E. Moore1 1Institute of Child Health, London WC1N 1EH, United Kingdom; 2Institute of Molecular Genetics, CNRS UMR-5535 and University of Montpellier-II, 34293 Montpellier, France; 3Sir William Dunn School of Pathology, University of Oxford, Oxford OX1 3RE, United Kingdom; 4Center for Applied Genomics, The Hospital for Sick Children, Toronto M5G 1L7, Canada; 5Inserm U741, F-75251 Paris Cedex 05, France Genomic imprinting is a developmentally important mechanism that involves both differential DNA methylation and allelic histone modifications. Through detailed comparative characterization, a large imprinted domain mapping to chromosome 7q21 in humans and proximal chromosome 6 in mice was redefined. This domain is organized around a maternally methylated CpG island comprising the promoters of the adjacent PEG10 and SGCE imprinted genes. Examination of Dnmt3l−/+ conceptuses shows that imprinted expression for all genes of the cluster depends upon the germline methylation at this putative “imprinting control region” (ICR). Similarly as for other ICRs, we find its DNA-methylated allele to be associated with trimethylation of lysine 9 on histone H3 (H3K9me3) and trimethylation of lysine 20 on histone H4 (H4K20me3), whereas the transcriptionally active paternal allele is enriched in H3K4me2 and H3K9 acetylation. -

Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for AP51634PU-N Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: WB Recommended Dilution: ELISA: 1/1000. Western Blot: 1/100-1/500. Reactivity: Human Host: Rabbit Isotype: Ig Clonality: Polyclonal Immunogen: KLH conjugated synthetic peptide between 191-221 amino acids from the Central region of Human FBXO5. Specificity: This antibody recognizes Human FBXO5 (Center). Formulation: PBS containing 0.09% (W/V) Sodium Azide as preservative State: Aff - Purified State: Liquid purified Ig fraction Concentration: lot specific Purification: Affinity Chromatography on Protein A Conjugation: Unconjugated Storage: Store undiluted at 2-8°C for one month or (in aliquots) at -20°C for longer. Avoid repeated freezing and thawing. Stability: Shelf life: one year from despatch. Gene Name: Homo sapiens F-box protein 5 (FBXO5), transcript variant 2 Database Link: Entrez Gene 26271 Human Q9UKT4 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Emi1 (FBXO5) (Center) Rabbit Polyclonal Antibody – AP51634PU-N Background: This gene encodes a member of the F-box protein family which is characterized by an approximately 40 amino acid motif, the F-box. The F-box proteins constitute one of the four subunits of the ubiquitin protein ligase complex called SCFs (SKP1-cullin-F-box), which function in phosphorylation-dependent ubiquitination. -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -

Mouse Ppp1r9a Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Ppp1r9a Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Ppp1r9a conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Ppp1r9a gene (NCBI Reference Sequence: NM_181595 ; Ensembl: ENSMUSG00000032827 ) is located on Mouse chromosome 6. 16 exons are identified, with the ATG start codon in exon 2 and the TGA stop codon in exon 16 (Transcript: ENSMUST00000035813). Exon 3 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Ppp1r9a gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-2F8 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a knock-out allele exhibit defects in dopamine-mediated neuromodulation, deficient long-term potentiation at corticostriatal synapses, increased spontaneous excitatory post-synaptic current frequency, and enhanced locomotor activationin response to cocaine treatment. Exon 3 starts from about 42.59% of the coding region. The knockout of Exon 3 will result in frameshift of the gene. The size of intron 2 for 5'-loxP site insertion: 139091 bp, and the size of intron 3 for 3'-loxP site insertion: 11389 bp. The size of effective cKO region: ~633 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 3 16 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Ppp1r9a Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Phosphorylation Sites in Protein Kinases and Phosphatases Regulated by Formyl Peptide Receptor 2 Signaling
International Journal of Molecular Sciences Review Phosphorylation Sites in Protein Kinases and Phosphatases Regulated by Formyl Peptide Receptor 2 Signaling Maria Carmela Annunziata 1, Melania Parisi 1, Gabriella Esposito 2 , Gabriella Fabbrocini 1, Rosario Ammendola 2 and Fabio Cattaneo 2,* 1 Department of Clinical Medicine and Surgery, School of Medicine, University of Naples Federico II, Via S. Pansini 5, 80131 Naples, Italy; [email protected] (M.C.A.); [email protected] (M.P.); [email protected] (G.F.) 2 Department of Molecular Medicine and Medical Biotechnology, School of Medicine,, University of Naples Federico II, Via S. Pansini 5, 80131 Naples, Italy; [email protected] (G.E.); [email protected] (R.A.) * Correspondence: [email protected]; Fax: +39-081-7464-359 Received: 5 May 2020; Accepted: 25 May 2020; Published: 27 May 2020 Abstract: FPR1, FPR2, and FPR3 are members of Formyl Peptides Receptors (FPRs) family belonging to the GPCR superfamily. FPR2 is a low affinity receptor for formyl peptides and it is considered the most promiscuous member of this family. Intracellular signaling cascades triggered by FPRs include the activation of different protein kinases and phosphatase, as well as tyrosine kinase receptors transactivation. Protein kinases and phosphatases act coordinately and any impairment of their activation or regulation represents one of the most common causes of several human diseases. Several phospho-sites has been identified in protein kinases and phosphatases, whose role may be to expand the repertoire of molecular mechanisms of regulation or may be necessary for fine-tuning of switch properties. We previously performed a phospho-proteomic analysis in FPR2-stimulated cells that revealed, among other things, not yet identified phospho-sites on six protein kinases and one protein phosphatase. -

Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications
Stem Cell Rev and Rep DOI 10.1007/s12015-016-9662-8 Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications Behnam Ahmadian Baghbaderani 1 & Adhikarla Syama2 & Renuka Sivapatham3 & Ying Pei4 & Odity Mukherjee2 & Thomas Fellner1 & Xianmin Zeng3,4 & Mahendra S. Rao5,6 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract We have recently described manufacturing of hu- help determine which set of tests will be most useful in mon- man induced pluripotent stem cells (iPSC) master cell banks itoring the cells and establishing criteria for discarding a line. (MCB) generated by a clinically compliant process using cord blood as a starting material (Baghbaderani et al. in Stem Cell Keywords Induced pluripotent stem cells . Embryonic stem Reports, 5(4), 647–659, 2015). In this manuscript, we de- cells . Manufacturing . cGMP . Consent . Markers scribe the detailed characterization of the two iPSC clones generated using this process, including whole genome se- quencing (WGS), microarray, and comparative genomic hy- Introduction bridization (aCGH) single nucleotide polymorphism (SNP) analysis. We compare their profiles with a proposed calibra- Induced pluripotent stem cells (iPSCs) are akin to embryonic tion material and with a reporter subclone and lines made by a stem cells (ESC) [2] in their developmental potential, but dif- similar process from different donors. We believe that iPSCs fer from ESC in the starting cell used and the requirement of a are likely to be used to make multiple clinical products. We set of proteins to induce pluripotency [3]. Although function- further believe that the lines used as input material will be used ally identical, iPSCs may differ from ESC in subtle ways, at different sites and, given their immortal status, will be used including in their epigenetic profile, exposure to the environ- for many years or even decades.