Marta Dora Phd Thesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cultivation-Independent Analysis of Pseudomonas Species in Soil and in the Rhizosphere of field-Grown Verticillium Dahliae Host Plants
Blackwell Publishing LtdOxford, UKEMIEnvironmental Microbiology1462-2912© 2006 The Authors; Journal compilation © 2006 Society for Applied Microbiology and Blackwell Publishing Ltd200681221362149Original Article Pseudomonas diversity in the rhizosphereR. Costa, J. F. Salles, G. Berg and K. Smalla Environmental Microbiology (2006) 8(12), 2136–2149 doi:10.1111/j.1462-2920.2006.01096.x Cultivation-independent analysis of Pseudomonas species in soil and in the rhizosphere of field-grown Verticillium dahliae host plants Rodrigo Costa,1 Joana Falcão Salles,2 Gabriele Berg3 rescens lineage and showed closest similarity to and Kornelia Smalla1* culturable Pseudomonas known for displaying anti- 1Federal Biological Research Centre for Agriculture and fungal properties. This report provides a better under- Forestry (BBA), Messeweg 11/12, D-38104 standing of how different factors drive Pseudomonas Braunschweig, Germany. community structure and diversity in bulk and rhizo- 2UMR 5557 Ecologie Microbienne (CNRS – Université sphere soils. Lyon 1), USC 1193 INRA, bâtiment G. Mendel, 43 boulevard du 11 Novembre 1918, F-69622 Villeurbanne, Introduction France. 3Graz University of Technology, Institute of Environmental Verticillium dahliae causes wilt of a broad range of crop Biotechnology, Petersgasse 12, A-8010 Graz, Austria. plants and significant annual yield losses worldwide (Tja- mos et al., 2000). Control of V. dahliae in soil had been largely dependent on the application of methyl bromide in Summary the field. As this ozone-depleting soil fumigant has been Despite their importance for rhizosphere functioning, recently phased-out, the use of alternative, ecologically rhizobacterial Pseudomonas spp. have been mainly friendly practices to combat V. dahliae is a subject of studied in a cultivation-based manner. -

Étude Des Communautés Microbiennes Rhizosphériques De Ligneux Indigènes De Sols Anthropogéniques, Issus D’Effluents Industriels Cyril Zappelini
Étude des communautés microbiennes rhizosphériques de ligneux indigènes de sols anthropogéniques, issus d’effluents industriels Cyril Zappelini To cite this version: Cyril Zappelini. Étude des communautés microbiennes rhizosphériques de ligneux indigènes de sols anthropogéniques, issus d’effluents industriels. Sciences agricoles. Université Bourgogne Franche- Comté, 2018. Français. NNT : 2018UBFCD057. tel-01902775 HAL Id: tel-01902775 https://tel.archives-ouvertes.fr/tel-01902775 Submitted on 23 Oct 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. UNIVERSITÉ DE BOURGOGNE FRANCHE-COMTÉ École doctorale Environnement-Santé Laboratoire Chrono-Environnement (UMR UFC/CNRS 6249) THÈSE Présentée en vue de l’obtention du titre de Docteur de l’Université Bourgogne Franche-Comté Spécialité « Sciences de la Vie et de l’Environnement » ÉTUDE DES COMMUNAUTES MICROBIENNES RHIZOSPHERIQUES DE LIGNEUX INDIGENES DE SOLS ANTHROPOGENIQUES, ISSUS D’EFFLUENTS INDUSTRIELS Présentée et soutenue publiquement par Cyril ZAPPELINI Le 3 juillet 2018, devant le jury composé de : Membres du jury : Vera SLAVEYKOVA (Professeure, Univ. de Genève) Rapporteure Bertrand AIGLE (Professeur, Univ. de Lorraine) Rapporteur & président du jury Céline ROOSE-AMSALEG (IGR, Univ. de Rennes) Examinatrice Karine JEZEQUEL (Maître de conférences, Univ. de Haute Alsace) Examinatrice Nicolas CAPELLI (Maître de conférences HDR, UBFC) Encadrant Christophe GUYEUX (Professeur, UBFC) Co-directeur de thèse Michel CHALOT (Professeur, UBFC) Directeur de thèse « En vérité, le chemin importe peu, la volonté d'arriver suffit à tout. -

Università Degli Studi Di Padova Dipartimento Di Biomedicina Comparata Ed Alimentazione
UNIVERSITÀ DEGLI STUDI DI PADOVA DIPARTIMENTO DI BIOMEDICINA COMPARATA ED ALIMENTAZIONE SCUOLA DI DOTTORATO IN SCIENZE VETERINARIE Curriculum Unico Ciclo XXVIII PhD Thesis INTO THE BLUE: Spoilage phenotypes of Pseudomonas fluorescens in food matrices Director of the School: Illustrious Professor Gianfranco Gabai Department of Comparative Biomedicine and Food Science Supervisor: Dr Barbara Cardazzo Department of Comparative Biomedicine and Food Science PhD Student: Andreani Nadia Andrea 1061930 Academic year 2015 To my family of origin and my family that is to be To my beloved uncle Piero Science needs freedom, and freedom presupposes responsibility… (Professor Gerhard Gottschalk, Göttingen, 30th September 2015, ProkaGENOMICS Conference) Table of Contents Table of Contents Table of Contents ..................................................................................................................... VII List of Tables............................................................................................................................. XI List of Illustrations ................................................................................................................ XIII ABSTRACT .............................................................................................................................. XV ESPOSIZIONE RIASSUNTIVA ............................................................................................ XVII ACKNOWLEDGEMENTS .................................................................................................... -

(12) United States Patent (10) Patent No.: US 7476,532 B2 Schneider Et Al
USOO7476532B2 (12) United States Patent (10) Patent No.: US 7476,532 B2 Schneider et al. (45) Date of Patent: Jan. 13, 2009 (54) MANNITOL INDUCED PROMOTER Makrides, S.C., "Strategies for achieving high-level expression of SYSTEMIS IN BACTERAL, HOST CELLS genes in Escherichia coli,” Microbiol. Rev. 60(3):512-538 (Sep. 1996). (75) Inventors: J. Carrie Schneider, San Diego, CA Sánchez-Romero, J., and De Lorenzo, V., "Genetic engineering of nonpathogenic Pseudomonas strains as biocatalysts for industrial (US); Bettina Rosner, San Diego, CA and environmental process.” in Manual of Industrial Microbiology (US) and Biotechnology, Demain, A, and Davies, J., eds. (ASM Press, Washington, D.C., 1999), pp. 460-474. (73) Assignee: Dow Global Technologies Inc., Schneider J.C., et al., “Auxotrophic markers pyrF and proC can Midland, MI (US) replace antibiotic markers on protein production plasmids in high cell-density Pseudomonas fluorescens fermentation.” Biotechnol. (*) Notice: Subject to any disclaimer, the term of this Prog., 21(2):343-8 (Mar.-Apr. 2005). patent is extended or adjusted under 35 Schweizer, H.P.. "Vectors to express foreign genes and techniques to U.S.C. 154(b) by 0 days. monitor gene expression in Pseudomonads. Curr: Opin. Biotechnol., 12(5):439-445 (Oct. 2001). (21) Appl. No.: 11/447,553 Slater, R., and Williams, R. “The expression of foreign DNA in bacteria.” in Molecular Biology and Biotechnology, Walker, J., and (22) Filed: Jun. 6, 2006 Rapley, R., eds. (The Royal Society of Chemistry, Cambridge, UK, 2000), pp. 125-154. (65) Prior Publication Data Stevens, R.C., “Design of high-throughput methods of protein pro duction for structural biology.” Structure, 8(9):R177-R185 (Sep. -

Aquatic Microbial Ecology 80:15
The following supplement accompanies the article Isolates as models to study bacterial ecophysiology and biogeochemistry Åke Hagström*, Farooq Azam, Carlo Berg, Ulla Li Zweifel *Corresponding author: [email protected] Aquatic Microbial Ecology 80: 15–27 (2017) Supplementary Materials & Methods The bacteria characterized in this study were collected from sites at three different sea areas; the Northern Baltic Sea (63°30’N, 19°48’E), Northwest Mediterranean Sea (43°41'N, 7°19'E) and Southern California Bight (32°53'N, 117°15'W). Seawater was spread onto Zobell agar plates or marine agar plates (DIFCO) and incubated at in situ temperature. Colonies were picked and plate- purified before being frozen in liquid medium with 20% glycerol. The collection represents aerobic heterotrophic bacteria from pelagic waters. Bacteria were grown in media according to their physiological needs of salinity. Isolates from the Baltic Sea were grown on Zobell media (ZoBELL, 1941) (800 ml filtered seawater from the Baltic, 200 ml Milli-Q water, 5g Bacto-peptone, 1g Bacto-yeast extract). Isolates from the Mediterranean Sea and the Southern California Bight were grown on marine agar or marine broth (DIFCO laboratories). The optimal temperature for growth was determined by growing each isolate in 4ml of appropriate media at 5, 10, 15, 20, 25, 30, 35, 40, 45 and 50o C with gentle shaking. Growth was measured by an increase in absorbance at 550nm. Statistical analyses The influence of temperature, geographical origin and taxonomic affiliation on growth rates was assessed by a two-way analysis of variance (ANOVA) in R (http://www.r-project.org/) and the “car” package. -

Evolutionary Divergence of the Wsp Signal Transduction System in Β- and Γ-Proteobacteria
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.02.450980; this version posted July 3, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Evolutionary divergence of the Wsp signal transduction system in β- and γ-proteobacteria 2 Collin Kessler1, Eisha Mhatre2, Vaughn Cooper2, and Wook Kim1* 3 4 1 Department of Biological Sciences, Duquesne University, Pittsburgh, 15282 5 2 Department of Microbiology and Molecular Genetics, University of Pittsburgh, Pittsburgh, 15219 6 *Correspondence: [email protected] 7 8 Abstract 9 Bacteria rapidly adapt to their environment by integrating external stimuli through diverse signal 10 transduction systems. Pseudomonas aeruginosa, for example, senses surface-contact through the Wsp 11 signal transduction system to trigger the production of cyclic di-GMP. Diverse mutations in wsp genes 12 that manifest enhanced biofilm formation are frequently reported in clinical isolates of P. aeruginosa, and 13 in biofilm studies of Pseudomonas spp. and Burkholderia cenocepacia. In contrast to the convergent 14 phenotypes associated with comparable wsp mutations, we demonstrate that the Wsp system in B. 15 cenocepacia does not impact intracellular cyclic di-GMP levels unlike that in Pseudomonas spp. Our 16 current mechanistic understanding of the Wsp system is entirely based on the study of four Pseudomonas 17 spp. and its phylogenetic distribution remains unknown. Here, we present the first broad phylogenetic 18 analysis to date to show that the Wsp system originated in the β-proteobacteria then horizontally 19 transferred to Pseudomonas spp., the sole member of the γ-proteobacteria. -

Control of Phytopathogenic Microorganisms with Pseudomonas Sp. and Substances and Compositions Derived Therefrom
(19) TZZ Z_Z_T (11) EP 2 820 140 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A01N 63/02 (2006.01) A01N 37/06 (2006.01) 10.01.2018 Bulletin 2018/02 A01N 37/36 (2006.01) A01N 43/08 (2006.01) C12P 1/04 (2006.01) (21) Application number: 13754767.5 (86) International application number: (22) Date of filing: 27.02.2013 PCT/US2013/028112 (87) International publication number: WO 2013/130680 (06.09.2013 Gazette 2013/36) (54) CONTROL OF PHYTOPATHOGENIC MICROORGANISMS WITH PSEUDOMONAS SP. AND SUBSTANCES AND COMPOSITIONS DERIVED THEREFROM BEKÄMPFUNG VON PHYTOPATHOGENEN MIKROORGANISMEN MIT PSEUDOMONAS SP. SOWIE DARAUS HERGESTELLTE SUBSTANZEN UND ZUSAMMENSETZUNGEN RÉGULATION DE MICRO-ORGANISMES PHYTOPATHOGÈNES PAR PSEUDOMONAS SP. ET DES SUBSTANCES ET DES COMPOSITIONS OBTENUES À PARTIR DE CELLE-CI (84) Designated Contracting States: • O. COUILLEROT ET AL: "Pseudomonas AL AT BE BG CH CY CZ DE DK EE ES FI FR GB fluorescens and closely-related fluorescent GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO pseudomonads as biocontrol agents of PL PT RO RS SE SI SK SM TR soil-borne phytopathogens", LETTERS IN APPLIED MICROBIOLOGY, vol. 48, no. 5, 1 May (30) Priority: 28.02.2012 US 201261604507 P 2009 (2009-05-01), pages 505-512, XP55202836, 30.07.2012 US 201261670624 P ISSN: 0266-8254, DOI: 10.1111/j.1472-765X.2009.02566.x (43) Date of publication of application: • GUANPENG GAO ET AL: "Effect of Biocontrol 07.01.2015 Bulletin 2015/02 Agent Pseudomonas fluorescens 2P24 on Soil Fungal Community in Cucumber Rhizosphere (73) Proprietor: Marrone Bio Innovations, Inc. -

Health Hazard Evaluation Report 2014-0050-3234
Evaluation of an Unpleasant Odor at an Aircraft Ejection Seat Manufacturer Kendra Broadwater, MPH Marie A. de Perio, MD Scott E. Brueck, MS, CIH Nancy C. Burton, PhD, CIH Angela R. Lemons, MS Brett J. Green, PhD HHE Report No. 2014-0050-3234 April 2015 U.S. Department of Health and Human Services Centers for Disease Control and Prevention Page 1 NationalHealth Institute Hazard forEvaluation Occupational Report 2014-0050-3234 Safety and Health Contents Highlights ...............................................i Abbreviations ..................................... iii Introduction ......................................... 1 Methods ............................................... 2 Results and Discussion ........................ 4 Conclusions ........................................ 14 Recommendations............................. 15 Appendix A ........................................ 18 Appendix B......................................... 22 Appendix C ........................................ 25 Appendix D ........................................27 References......................................... 28 Acknowledgement............................33 The employer is required to post a copy of this report for 30 days at or near the workplace(s) of affected employees. The employer must take steps to ensure that the posted report is not altered, defaced, or covered by other material. The cover photo is a close-up image of sorbent tubes, which are used by the HHE Program to measure airborne exposures. This photo is an artistic representation that may not be related to this Health Hazard Evaluation. Photo by NIOSH. Page 2 Health Hazard Evaluation Report 2014-0050-3234 Highlights of this Evaluation The Health Hazard Evaluation Program received a request from the employer at a facility where aircraft ejection seats are made. Employees were concerned about a lingering odor that was first noticed in April 2013 and continued to be a problem. The odor remained in materials and goods after they left the facility. What We Did ● We visited the facility in May 2014. -
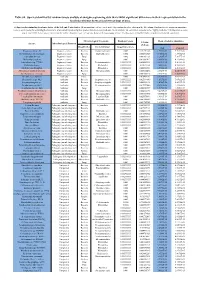
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

Pseudomonas Versuta Sp. Nov., Isolated from Antarctic Soil 1 Wah
*Manuscript 1 Pseudomonas versuta sp. nov., isolated from Antarctic soil 1 2 3 1,2 3 1 2,4 1,5 4 2 Wah Seng See-Too , Sergio Salazar , Robson Ee , Peter Convey , Kok-Gan Chan , 5 6 3 Álvaro Peix 3,6* 7 8 4 1Division of Genetics and Molecular Biology, Institute of Biological Sciences, Faculty of 9 10 11 5 Science University of Malaya, 50603 Kuala Lumpur, Malaysia 12 13 6 2National Antarctic Research Centre (NARC), Institute of Postgraduate Studies, University of 14 15 16 7 Malaya, 50603 Kuala Lumpur, Malaysia 17 18 8 3Instituto de Recursos Naturales y Agrobiología. IRNASA -CSIC, Salamanca, Spain 19 20 4 21 9 British Antarctic Survey, NERC, High Cross, Madingley Road, Cambridge CB3 OET, UK 22 23 10 5UM Omics Centre, University of Malaya, Kuala Lumpur, Malaysia 24 25 11 6Unidad Asociada Grupo de Interacción Planta-Microorganismo Universidad de Salamanca- 26 27 28 12 IRNASA ( CSIC) 29 30 13 , IRNASA-CSIC, 31 32 33 14 c/Cordel de Merinas 40 -52, 37008 Salamanca, Spain. Tel.: +34 923219606. 34 35 15 E-mail address: [email protected] (A. Peix) 36 37 38 39 16 Abstract: 40 41 42 43 17 In this study w e used a polyphas ic taxonomy approach to analyse three bacterial strains 44 45 18 coded L10.10 T, A4R1.5 and A4R1.12 , isolated in the course of a study of quorum -quenching 46 47 19 bacteria occurring Antarctic soil . The 16S rRNA gene sequence was identical in the three 48 49 50 20 strains and showed 99.7% pairwise similarity with respect to the closest related species 51 52 21 Pseudomonas weihenstephanensis WS4993 T, and the next closest related species were P. -

Molecular Analysis of Poultry Meat Spoiling Microbiota and Heterogeneity of Their Proteolytic and Lipolytic Enzyme Activities
Acta Alimentaria, Vol. 40 (Suppl.), pp. 3–22 (2011) DOI: 10.1556/AAlim.40.2011.Suppl.2 MOLECULAR ANALYSIS OF POULTRY MEAT SPOILING MICROBIOTA AND HETEROGENEITY OF THEIR PROTEOLYTIC AND LIPOLYTIC ENZYME ACTIVITIES Á. BELÁKa, M. KOVÁCSa, ZS. HERMANNa, Á. N. HOLCZMANa, D. MÁRTAa, S. CENIČ STOJAKOVIČb, N. BAJCSIa and A. MARÁZa* aDepartment of Microbiology and Biotechnology, Faculty of Food Science, Corvinus University of Budapest, H-1118 Budapest, Somlói út 14–16. Hungary bDepartment of Chemistry and Biochemistry, Faculty of Food Technology and Biotechnology, University of Zagreb, Pierottijeva 6, 10000 Zagreb. Croatia Psychrotrophic Pseudomonas species P. fl uorescens, P. fragi and P. lundensis were found as predominant bacteria of chicken meat stored at chill temperature, which showed high level of molecular diversity, while isolates of the psychrotrophic yeasts Candida zeylanoides, Metschnikowia pulcherrima, Rhodotorula glutinis and Rhodotorula mucilaginosa formed clusters of high level similarity within the different species as revealed by RAPD-PCR analysis. Combination of multiplex PCR and sequencing of the rpoB gene resulted correct identifi cation of the Pseudomas isolates, while the routine diagnostic tests led to improper identifi cation in case of half of the isolates, which indicated the extended biochemical and physiological heterogeneity of the food-borne pseudomonads. Majority of P. fl uorescens and P. lundensis isolates were strong protease and lipase producers, while P. fragi strains were week or negative from this respect. Proteolytic and lipolytic activities of the isolated yeast strains were species specifi c and protease production was less frequent than lipolytic activities. Keywords: poultry meat, spoilage, Pseudomonas, yeasts, molecular characterisation, protease, lipase Meat spoilage is determined by numerous environmental factors, although the metabolic activity of microorganisms plays the most important role. -
Pseudomonas Versuta Sp. Nov., Isolated from Antarctic Soil
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by NERC Open Research Archive Accepted Manuscript Title: Pseudomonas versuta sp. nov., isolated from Antarctic soil Authors: Wah Seng See-Too, Sergio Salazar, Robson Ee, Peter Convey, Kok-Gan Chan, Alvaro´ Peix PII: S0723-2020(17)30039-5 DOI: http://dx.doi.org/doi:10.1016/j.syapm.2017.03.002 Reference: SYAPM 25827 To appear in: Received date: 12-1-2017 Revised date: 20-3-2017 Accepted date: 24-3-2017 Please cite this article as: Wah Seng See-Too, Sergio Salazar, Robson Ee, Peter Convey, Kok-Gan Chan, Alvaro´ Peix, Pseudomonas versuta sp.nov., isolated from Antarctic soil, Systematic and Applied Microbiologyhttp://dx.doi.org/10.1016/j.syapm.2017.03.002 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. Pseudomonas versuta sp. nov., isolated from Antarctic soil Wah Seng See-Too1,2, Sergio Salazar3, Robson Ee1, Peter Convey 2,4, Kok-Gan Chan1,5, Álvaro Peix3,6* 1Division of Genetics and Molecular Biology, Institute of Biological Sciences, Faculty of Science University of Malaya, 50603 Kuala Lumpur, Malaysia 2National Antarctic Research Centre (NARC), Institute of Postgraduate Studies, University of Malaya, 50603 Kuala Lumpur, Malaysia 3Instituto de Recursos Naturales y Agrobiología.