Radiation Response and Regulation of Apoptosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetics of Interleukin 1 Receptor-Like 1 in Immune and Inflammatory Diseases
Current Genomics, 2010, 11, 591-606 591 Genetics of Interleukin 1 Receptor-Like 1 in Immune and Inflammatory Diseases Loubna Akhabir and Andrew Sandford* Department of Medicine, University of British Columbia, UBC James Hogg Research Centre, Providence Heart + Lung Institute, Room 166, St. Paul's Hospital, 1081 Burrard Street, Vancouver, BC V6Z 1Y6, Canada Abstract: Interleukin 1 receptor-like 1 (IL1RL1) is gaining in recognition due to its involvement in immune/inflamma- tory disorders. Well-designed animal studies have shown its critical role in experimental allergic inflammation and human in vitro studies have consistently demonstrated its up-regulation in several conditions such as asthma and rheumatoid ar- thritis. The ligand for IL1RL1 is IL33 which emerged as playing an important role in initiating eosinophilic inflammation and activating other immune cells resulting in an allergic phenotype. An IL1RL1 single nucleotide polymorphism (SNP) was among the most significant results of a genome-wide scan inves- tigating eosinophil counts; in the same study, this SNP associated with asthma in 10 populations. The IL1RL1 gene resides in a region of high linkage disequilibrium containing interleukin 1 receptor genes as well as in- terleukin 18 receptor and accessory genes. This poses a challenge to researchers interested in deciphering genetic associa- tion signals in the region as all of the genes represent interesting candidates for asthma and allergic disease. The IL1RL1 gene and its resulting soluble and receptor proteins have emerged as key regulators of the inflammatory proc- ess implicated in a large variety of human pathologies We review the function and expression of the IL1RL1 gene. -

Oncostatin M Exhibit Elevated Responsiveness to IL-31 Receptor
IL-31 Receptor (IL-31RA) Knockout Mice Exhibit Elevated Responsiveness to Oncostatin M This information is current as Janine Bilsborough, Sherri Mudri, Eric Chadwick, Brandon of September 28, 2021. Harder and Stacey R. Dillon J Immunol 2010; 185:6023-6030; Prepublished online 18 October 2010; doi: 10.4049/jimmunol.0902769 http://www.jimmunol.org/content/185/10/6023 Downloaded from References This article cites 29 articles, 6 of which you can access for free at: http://www.jimmunol.org/content/185/10/6023.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2010 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology IL-31 Receptor (IL-31RA) Knockout Mice Exhibit Elevated Responsiveness to Oncostatin M Janine Bilsborough,1 Sherri Mudri,1 Eric Chadwick,2 Brandon Harder,3 and Stacey R. Dillon IL-31 signals through the heterodimeric receptor IL-31RA and oncostatin M receptor (OSMR), and has been linked with the development of atopic dermatitis, a Th2 cytokine-associated disease in humans. -

System, Method and Software for Calculation of a Cannabis Drug Efficiency Index for the Reduction of Inflammation
International Journal of Molecular Sciences Article System, Method and Software for Calculation of a Cannabis Drug Efficiency Index for the Reduction of Inflammation Nicolas Borisov 1,† , Yaroslav Ilnytskyy 2,3,†, Boseon Byeon 2,3,4,†, Olga Kovalchuk 2,3 and Igor Kovalchuk 2,3,* 1 Moscow Institute of Physics and Technology, 9 Institutsky lane, Dolgoprudny, Moscow Region 141701, Russia; [email protected] 2 Department of Biological Sciences, University of Lethbridge, Lethbridge, AB T1K 3M4, Canada; [email protected] (Y.I.); [email protected] (B.B.); [email protected] (O.K.) 3 Pathway Rx., 16 Sandstone Rd. S., Lethbridge, AB T1K 7X8, Canada 4 Biomedical and Health Informatics, Computer Science Department, State University of New York, 2 S Clinton St, Syracuse, NY 13202, USA * Correspondence: [email protected] † First three authors contributed equally to this research. Abstract: There are many varieties of Cannabis sativa that differ from each other by composition of cannabinoids, terpenes and other molecules. The medicinal properties of these cultivars are often very different, with some being more efficient than others. This report describes the development of a method and software for the analysis of the efficiency of various cannabis extracts to detect the anti-inflammatory properties of the various cannabis extracts. The method uses high-throughput gene expression profiling data but can potentially use other omics data as well. According to the signaling pathway topology, the gene expression profiles are convoluted into the signaling pathway activities using a signaling pathway impact analysis (SPIA) method. The method was tested by inducing inflammation in human 3D epithelial tissues, including intestine, oral and skin, and then exposing these tissues to various extracts and then performing transcriptome analysis. -

How Relevant Are Bone Marrow-Derived Mast Cells (Bmmcs) As Models for Tissue Mast Cells? a Comparative Transcriptome Analysis of Bmmcs and Peritoneal Mast Cells
cells Article How Relevant Are Bone Marrow-Derived Mast Cells (BMMCs) as Models for Tissue Mast Cells? A Comparative Transcriptome Analysis of BMMCs and Peritoneal Mast Cells 1, 2, 1 1 2,3 Srinivas Akula y , Aida Paivandy y, Zhirong Fu , Michael Thorpe , Gunnar Pejler and Lars Hellman 1,* 1 Department of Cell and Molecular Biology, Uppsala University, The Biomedical Center, Box 596, SE-751 24 Uppsala, Sweden; [email protected] (S.A.); [email protected] (Z.F.); [email protected] (M.T.) 2 Department of Medical Biochemistry and Microbiology, Uppsala University, The Biomedical Center, Box 589, SE-751 23 Uppsala, Sweden; [email protected] (A.P.); [email protected] (G.P.) 3 Department of Anatomy, Physiology and Biochemistry, Swedish University of Agricultural Sciences, Box 7011, SE-75007 Uppsala, Sweden * Correspondence: [email protected]; Tel.: +46-(0)18-471-4532; Fax: +46-(0)18-471-4862 These authors contributed equally to this work. y Received: 29 July 2020; Accepted: 16 September 2020; Published: 17 September 2020 Abstract: Bone marrow-derived mast cells (BMMCs) are often used as a model system for studies of the role of MCs in health and disease. These cells are relatively easy to obtain from total bone marrow cells by culturing under the influence of IL-3 or stem cell factor (SCF). After 3 to 4 weeks in culture, a nearly homogenous cell population of toluidine blue-positive cells are often obtained. However, the question is how relevant equivalents these cells are to normal tissue MCs. By comparing the total transcriptome of purified peritoneal MCs with BMMCs, here we obtained a comparative view of these cells. -

The Interleukin-18 Receptor Is Differentially Expressed in Whole
1 The interleukin-18 receptor is differentially expressed in the blood during sepsis. 2 3 Shahan Mamoor, MS1 4 [email protected] East Islip, NY, USA 5 6 We probed published and public microarray datasets1,2 to discover the most significant gene expression changes in the blood of patients with sepsis. We found significant induction 7 expression of IL18RAP and IL18R1, genes encoding subunits of the interleukin-18 receptor, in 8 whole blood from patients with sepsis. 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 Keywords: sepsis, septic shock, interleukin-18, IL18R1, IL18RAP, systems biology of septic 26 shock. 27 28 PAGE 1 OF 13 1 Septic shock is a leading cause of mortality in the United States and worldwide3. We 2 used published and public microarray datasets1,2 to identify differentially expressed genes in the 3 4 blood of patients with sepsis. We identified IL18R1 and IL18RAP as among the genes most 5 differentially expressed in blood in the septic state. 6 7 Methods 8 9 We utilized microarray datasets GSE1001591 and GSE264402 for this differential gene 10 11 expression analysis of blood cells during sepsis. GSE100509 was generated with whole blood 12 using Illumina HumanWG-6 v3.0 expression beadchip technology with n=12 whole blood from 13 control subjects and n=33 whole blood from sepsis patients. GSE26440 was generated using 14 15 Affymetrix Human Genome U133 Plus 2.0 Array technology with n=32 control subjects and 16 n=98 sepsis patients. The Benjamini and Hochberg method of p-value adjustment was used for 17 ranking of differential expression but raw p-values were used for assessment of statistical 18 19 significance of global differential expression. -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

IL-31 Uncouples Skin Inflammation from Itch Sensation in Allergic Dermatitis
UCSF UC San Francisco Previously Published Works Title IL-31 uncouples skin inflammation from itch sensation in allergic dermatitis Permalink https://escholarship.org/uc/item/9sh4x406 Authors Fassett, Marlys S Braz, Joao M Castellanos, Carlos A et al. Publication Date 2021-05-13 DOI 10.1101/2021.05.12.443916 Peer reviewed eScholarship.org Powered by the California Digital Library University of California bioRxiv preprint doi: https://doi.org/10.1101/2021.05.12.443916; this version posted May 13, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: IL-31 uncouples skin inflammation from itch sensation in allergic dermatitis Authors: Marlys S. Fassett,1,2,3 Joao M. Braz,4 Carlos A. Castellanos,2,3 Andrew W. Schroeder,3 Mahsa Sadeghi,4 Darryl J. Mar,2 Connie J. Zhou,2 Jeoung-Sook Shin,2,3 Allan I. Basbaum,4 and K. Mark Ansel2,3 Affiliations: 1Department of Dermatology, University of California, San Francisco; San Francisco, USA 2Department of Microbiology and Immunology, University of California, San Francisco; San Francisco, USA. 3Sandler Asthma Basic Research Center (SABRe); San Francisco, USA. 4Department of Anatomy, University of California San Francisco; San Francisco, USA. *Corresponding author. Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.05.12.443916; this version posted May 13, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Despite a robust literature associating IL-31 with pruritic inflammatory skin diseases, its influence on cutaneous inflammation and on the interplay between inflammatory and neurosensory pathways remain unmapped. -
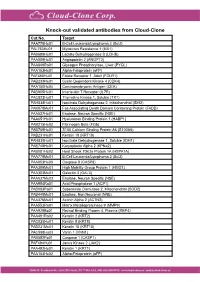
Knock-Out Validated Antibodies from Cloud-Clone Cat.No
Knock-out validated antibodies from Cloud-Clone Cat.No. Target PAA778Hu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAL763Hu01 Myxovirus Resistance 1 (MX1) PAB698Hu01 Lactate Dehydrogenase B (LDHB) PAA009Hu01 Angiopoietin 2 (ANGPT2) PAA849Ra01 Glycogen Phosphorylase, Liver (PYGL) PAA153Hu01 Alpha-Fetoprotein (aFP) PAF460Hu01 Folate Receptor 1, Adult (FOLR1) PAB233Hu01 Cyclin Dependent Kinase 4 (CDK4) PAA150Hu04 Carcinoembryonic Antigen (CEA) PAB905Hu01 Interleukin 7 Receptor (IL7R) PAC823Hu01 Thymidine Kinase 1, Soluble (TK1) PAH838Hu01 Isocitrate Dehydrogenase 2, mitochondrial (IDH2) PAK078Mu01 Fas Associating Death Domain Containing Protein (FADD) PAA537Hu01 Enolase, Neuron Specific (NSE) PAA651Hu01 Hyaluronan Binding Protein 1 (HABP1) PAB215Hu02 Fibrinogen Beta (FGb) PAB769Hu01 S100 Calcium Binding Protein A6 (S100A6) PAB231Hu01 Keratin 18 (KRT18) PAH839Hu01 Isocitrate Dehydrogenase 1, Soluble (IDH1) PAE748Hu01 Karyopherin Alpha 2 (KPNa2) PAB081Hu02 Heat Shock 70kDa Protein 1A (HSPA1A) PAA778Mu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAA853Hu03 Caspase 8 (CASP8) PAA399Mu01 High Mobility Group Protein 1 (HMG1) PAA303Mu01 Galectin 3 (GAL3) PAA537Mu02 Enolase, Neuron Specific (NSE) PAA994Ra01 Acid Phosphatase 1 (ACP1) PAB083Ra01 Superoxide Dismutase 2, Mitochondrial (SOD2) PAB449Mu01 Enolase, Non Neuronal (NNE) PAA376Mu01 Actinin Alpha 2 (ACTN2) PAA553Ra01 Matrix Metalloproteinase 9 (MMP9) PAA929Bo01 Retinol Binding Protein 4, Plasma (RBP4) PAA491Ra02 Keratin 2 (KRT2) PAC025Hu01 Keratin 8 (KRT8) PAB231Mu01 Keratin 18 (KRT18) PAC598Hu03 Vanin 1 (VNN1) -

Evolutionary Divergence and Functions of the Human Interleukin (IL) Gene Family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W
UPDATE ON GENE COMPLETIONS AND ANNOTATIONS Evolutionary divergence and functions of the human interleukin (IL) gene family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W. Nebert3* and Vasilis Vasiliou1 1Molecular Toxicology and Environmental Health Sciences Program, Department of Pharmaceutical Sciences, University of Colorado Denver, Aurora, CO 80045, USA 2Department of Clinical Pharmacy, University of Colorado Denver, Aurora, CO 80045, USA 3Department of Environmental Health and Center for Environmental Genetics (CEG), University of Cincinnati Medical Center, Cincinnati, OH 45267–0056, USA *Correspondence to: Tel: þ1 513 821 4664; Fax: þ1 513 558 0925; E-mail: [email protected]; [email protected] Date received (in revised form): 22nd September 2010 Abstract Cytokines play a very important role in nearly all aspects of inflammation and immunity. The term ‘interleukin’ (IL) has been used to describe a group of cytokines with complex immunomodulatory functions — including cell proliferation, maturation, migration and adhesion. These cytokines also play an important role in immune cell differentiation and activation. Determining the exact function of a particular cytokine is complicated by the influence of the producing cell type, the responding cell type and the phase of the immune response. ILs can also have pro- and anti-inflammatory effects, further complicating their characterisation. These molecules are under constant pressure to evolve due to continual competition between the host’s immune system and infecting organisms; as such, ILs have undergone significant evolution. This has resulted in little amino acid conservation between orthologous proteins, which further complicates the gene family organisation. Within the literature there are a number of overlapping nomenclature and classification systems derived from biological function, receptor-binding properties and originating cell type. -

Estrogen Receptor Alpha (ESR1)-Dependent Regulation of the Mouse Oviductal Transcriptome Katheryn L
University of Kentucky UKnowledge Animal and Food Sciences Faculty Publications Animal and Food Sciences 1-25-2016 Estrogen Receptor Alpha (ESR1)-Dependent Regulation of the Mouse Oviductal Transcriptome Katheryn L. Cerny University of Kentucky, [email protected] Rosanne A. C. Ribeiro University of Kentucky Myoungkun Jeoung University of Kentucky, [email protected] CheMyong Ko University of Illinois at Urbana-Champaign Phillip J. Bridges University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits oy u. Follow this and additional works at: https://uknowledge.uky.edu/animalsci_facpub Part of the Animal Sciences Commons, Cell and Developmental Biology Commons, Food Science Commons, and the Physiology Commons Repository Citation Cerny, Katheryn L.; Ribeiro, Rosanne A. C.; Jeoung, Myoungkun; Ko, CheMyong; and Bridges, Phillip J., "Estrogen Receptor Alpha (ESR1)-Dependent Regulation of the Mouse Oviductal Transcriptome" (2016). Animal and Food Sciences Faculty Publications. 7. https://uknowledge.uky.edu/animalsci_facpub/7 This Article is brought to you for free and open access by the Animal and Food Sciences at UKnowledge. It has been accepted for inclusion in Animal and Food Sciences Faculty Publications by an authorized administrator of UKnowledge. For more information, please contact [email protected]. Estrogen Receptor Alpha (ESR1)-Dependent Regulation of the Mouse Oviductal Transcriptome Notes/Citation Information Published in PLOS ONE, v. 11, no. 1, e0147685, p. 1-17. © 2016 Cerny et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. -

Differentially Methylated Genes
10/30/2013 Disclosures Key Rheumatoid Arthritis-Associated Pathogenic Pathways Revealed by Integrative Analysis of RA Omics Datasets Consultant: IGNYTA Funding: Rheumatology Research Foundation By John W. Whitaker, Wei Wang and Gary S. Firestein DNA methylation and gene regulation The RA methylation signature in FLS DNA methylation – DNMT1 (maintaining methylation) OA – DNMT3a, 3b (de novo methylation) RA % of CpG methylation: 0% 100% Nakano et al. 2013 ARD AA06 AANAT AARS ABCA6 ABCC12 ABCG1 ABHD8 ABL2 ABR ABRA ACACA ACAN ACAP3 ACCSL ACN9 ACOT7 ACOX2 ACP5 ACP6 ACPP ACSL1 ACSL3 ACSM5 ACVRL1 ADAM10 ADAM32 ADAM33 ADAMTS12 ADAMTS15 ADAMTS19 ADAMTS4 ADAT3 ADCK4 ADCK5 ADCY2 ADCY3 ADCY6 ADORA1 ADPGK ADPRHL1 ADTRP AFAP1 AFAP1L2 AFF3 AFG3L1P AGAP11 AGER AGTR1 AGXT AIF1L AIM2 AIRE AJUBA AK4 AKAP12 AKAP2 AKR1C2 AKR1E2 AKT2 ALAS1 ALDH1L1-AS1 ALDH3A1 ALDH3B1 ALDH8A1 ALDOB ALDOC ALOX12 ALPK3 ALS2CL ALX4 AMBRA1 AMPD2 AMPD3 ANGPT1 ANGPT2 ANGPTL5 ANGPTL6 ANK1 ANKMY2 ANKRD29 ANKRD37 ANKRD53 ANO3 ANO6 ANO7 ANP32C ANXA6 ANXA8L2 AP1G1 AP2A2 AP2M1 AP5B1 APBA2 APC APCDD1 APOBEC3B APOBEC3G APOC1 APOH APOL6 APOLD1 APOM AQP1 AQP10 AQP6 AQP9 ARAP1 ARHGAP24 ARHGAP42 ARHGEF19 ARHGEF25 ARHGEF3 ARHGEF37 ARHGEF7 ARL4C ARL6IP 5 ARL8B ARMC3 ARNTL2 ARPP21 ARRB1 ARSI ASAH2B ASB10 ASB2 ASCL2 ASIC4 ASPH ATF3 ATF7 ATL1 ATL3 ATP10A ATP1A1 ATP1A4 ATP2C1 ATP5A1 ATP5EP2 ATP5L2 ATP6V0CP3 ATP6V1C1 ATP6V1E2 ATXN7L1 ATXN7L2 AVPI1 AXIN2 B3GNT7 B3GNT8 B3GNTL1 BACH1 BAG3 Differential methylated genes in RA FLS BAIAP2L2 BANP BATF BATF2 BBS2 BCAS4 BCAT1 BCL7C BDKRB2 BEGAIN BEST1 BEST3 -

Human Colon Cancer Primer Library
Human Colon Cancer Primer Library Catalog No: HCCR-1 Supplier: RealTimePrimers Lot No: XXXXX Supplied as: solid Stability: store at -20°C Description Contains 88 primer sets directed against cytokine and chemokine receptor genes and 8 housekeeping gene primer sets. Provided in a 96-well microplate (20 ul - 10 uM). Perform up to 100 PCR arrays (based on 20 ul assay volume per reaction). Just add cDNA template and SYBR green master mix. Gene List: • CCR1 chemokine (C-C motif) receptor 1 • IL11RA interleukin 11 receptor, alpha • CCR2 chemokine (C-C motif) receptor 2 • IL11RB interleukin 11 receptor, beta • CCR3 chemokine (C-C motif) receptor 3 • IL12RB1 interleukin 12 receptor, beta 1 • CCR4 chemokine (C-C motif) receptor 4 • IL12RB2 interleukin 12 receptor, beta 2 • CCR5 chemokine (C-C motif) receptor 5 • IL13RA1 interleukin 13 receptor, alpha 1 • CCR6 chemokine (C-C motif) receptor 6 • IL13RA2 interleukin 13 receptor, alpha 2 • CCR7 chemokine (C-C motif) receptor 7 • IL15RA interleukin 15 receptor, alpha • CCR8 chemokine (C-C motif) receptor 8 • IL15RB interleukin 15 receptor, beta • CCR9 chemokine (C-C motif) receptor 9 • IL17RA interleukin 17 receptor A • CCR10 chemokine (C-C motif) receptor 10 • IL17RB interleukin 17 receptor B • CX3CR1 chemokine (C-X3-C motif) receptor 1 • IL17RC interleukin 17 receptor C • CXCR1 chemokine (C-X-C motif) receptor 1 • IL17RD interleukin 17 receptor D • CXCR2 chemokine (C-X-C motif) receptor 2 • IL17RE interleukin 17 receptor E • CXCR3 chemokine (C-X-C motif) receptor 3 • IL18R1 interleukin 18 receptor