Cytological Assessments and Transcriptome Profiling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Materials Evodiamine Inhibits Both Stem Cell and Non-Stem
Supplementary materials Evodiamine inhibits both stem cell and non-stem-cell populations in human cancer cells by targeting heat shock protein 70 Seung Yeob Hyun, Huong Thuy Le, Hye-Young Min, Honglan Pei, Yijae Lim, Injae Song, Yen T. K. Nguyen, Suckchang Hong, Byung Woo Han, Ho-Young Lee - 1 - Table S1. Short tandem repeat (STR) DNA profiles for human cancer cell lines used in this study. MDA-MB-231 Marker H1299 H460 A549 HCT116 (MDA231) Amelogenin XX XY XY XX XX D8S1179 10, 13 12 13, 14 10, 14, 15 13 D21S11 32.2 30 29 29, 30 30, 33.2 D7S820 10 9, 12 8, 11 11, 12 8 CSF1PO 12 11, 12 10, 12 7, 10 12, 13 D3S1358 17 15, 18 16 12, 16, 17 16 TH01 6, 9.3 9.3 8, 9.3 8, 9 7, 9.3 D13S317 12 13 11 10, 12 13 D16S539 12, 13 9 11, 12 11, 13 12 D2S1338 23, 24 17, 25 24 16 21 D19S433 14 14 13 11, 12 11, 14 vWA 16, 18 17 14 17, 22 15 TPOX 8 8 8, 11 8, 9 8, 9 D18S51 16 13, 15 14, 17 15, 17 11, 16 D5S818 11 9, 10 11 10, 11 12 FGA 20 21, 23 23 18, 23 22, 23 - 2 - Table S2. Antibodies used in this study. Catalogue Target Vendor Clone Dilution ratio Application1) Number 1:1000 (WB) ADI-SPA- 1:50 (IHC) HSP70 Enzo C92F3A-5 WB, IHC, IF, IP 810-F 1:50 (IF) 1 :1000 (IP) ADI-SPA- HSP90 Enzo 9D2 1:1000 WB 840-F 1:1000 (WB) Oct4 Abcam ab19857 WB, IF 1:100 (IF) Nanog Cell Signaling 4903S D73G4 1:1000 WB Sox2 Abcam ab97959 1:1000 WB ADI-SRA- Hop Enzo DS14F5 1:1000 WB 1500-F HIF-1α BD 610958 54/HIF-1α 1:1000 WB pAkt (S473) Cell Signaling 4060S D9E 1:1000 WB Akt Cell Signaling 9272S 1:1000 WB pMEK Cell Signaling 9121S 1:1000 WB (S217/221) MEK Cell Signaling 9122S 1:1000 -

Note: the Letters 'F' and 'T' Following the Locators Refers to Figures and Tables
Index Note: The letters ‘f’ and ‘t’ following the locators refers to figures and tables cited in the text. A Acyl-lipid desaturas, 455 AA, see Arachidonic acid (AA) Adenophostin A, 71, 72t aa, see Amino acid (aa) Adenosine 5-diphosphoribose, 65, 789 AACOCF3, see Arachidonyl trifluoromethyl Adlea, 651 ketone (AACOCF3) ADP, 4t, 10, 155, 597, 598f, 599, 602, 669, α1A-adrenoceptor antagonist prazosin, 711t, 814–815, 890 553 ADPKD, see Autosomal dominant polycystic aa 723–928 fragment, 19 kidney disease (ADPKD) aa 839–873 fragment, 17, 19 ADPKD-causing mutations Aβ, see Amyloid β-peptide (Aβ) PKD1 ABC protein, see ATP-binding cassette protein L4224P, 17 (ABC transporter) R4227X, 17 Abeele, F. V., 715 TRPP2 Abbott Laboratories, 645 E837X, 17 ACA, see N-(p-amylcinnamoyl)anthranilic R742X, 17 acid (ACA) R807X, 17 Acetaldehyde, 68t, 69 R872X, 17 Acetic acid-induced nociceptive response, ADPR, see ADP-ribose (ADPR) 50 ADP-ribose (ADPR), 99, 112–113, 113f, Acetylcholine-secreting sympathetic neuron, 380–382, 464, 534–536, 535f, 179 537f, 538, 711t, 712–713, Acetylsalicylic acid, 49t, 55 717, 770, 784, 789, 816–820, Acrolein, 67t, 69, 867, 971–972 885 Acrosome reaction, 125, 130, 301, 325, β-Adrenergic agonists, 740 578, 881–882, 885, 888–889, α2 Adrenoreceptor, 49t, 55, 188 891–895 Adult polycystic kidney disease (ADPKD), Actinopterigy, 223 1023 Activation gate, 485–486 Aframomum daniellii (aframodial), 46t, 52 Leu681, amino acid residue, 485–486 Aframomum melegueta (Melegueta pepper), Tyr671, ion pathway, 486 45t, 51, 70 Acute myeloid leukaemia and myelodysplastic Agelenopsis aperta (American funnel web syndrome (AML/MDS), 949 spider), 48t, 54 Acylated phloroglucinol hyperforin, 71 Agonist-dependent vasorelaxation, 378 Acylation, 96 Ahern, G. -

Research Article Evodiamine Induces Transient Receptor Potential Vanilloid-1-Mediated Protective Autophagy in U87-MG Astrocytes
Hindawi Publishing Corporation Evidence-Based Complementary and Alternative Medicine Volume 2013, Article ID 354840, 9 pages http://dx.doi.org/10.1155/2013/354840 Research Article Evodiamine Induces Transient Receptor Potential Vanilloid-1-Mediated Protective Autophagy in U87-MG Astrocytes Ann-Jeng Liu,1,2 Sheng-Hao Wang,3 Sz-Ying Hou,3,4 Chien-Ju Lin,3 Wen-Ta Chiu,1,5 Sheng-Huang Hsiao,2 Thay-Hsiung Chen,6,7 and Chwen-Ming Shih3,4,8 1 Graduate Institute of Clinical Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan 2 Department of Neurosurgery, Taipei City Hospital Ren-Ai Branch, Taipei, Taiwan 3 Department of Biochemistry, School of Medicine, Taipei Medical University, 250 Wu-Hsing Street, Taipei 110, Taiwan 4 Graduate Institute of Medical Sciences, College of Medicine, Taipei Medical University, Taipei, Taiwan 5 Department of Neurosurgery, Taipei Municipal Wan-Fang Hospital, Taipei, Taiwan 6 Department of Surgery, College of Medicine, Taipei Medical University, Taiwan 7 Division of Cardiac Surgery, Cathy General Hospital, Taipei, Taiwan 8 Traditional Herbal Medicine Research Center, Taipei Medical University Hospital, Taipei, Taiwan Correspondence should be addressed to Thay-Hsiung Chen; [email protected] and Chwen-Ming Shih; [email protected] Received 21 October 2013; Accepted 23 November 2013 Academic Editor: Joen-Rong Sheu Copyright © 2013 Ann-Jeng Liu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Cerebral ischemia is a leading cause of mortality and morbidity worldwide, which results in cognitive and motor dysfunction, neurodegenerative diseases, and death. -
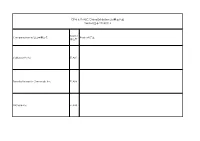
Cphi & P-MEC China Exhibition List展商名单version版本20180116
CPhI & P-MEC China Exhibition List展商名单 Version版本 20180116 Booth/ Company Name/公司中英文名 Product/产品 展位号 Carbosynth Ltd E1A01 Toronto Research Chemicals Inc E1A08 SiliCycle Inc. E1A10 SA TOURNAIRE E1A11 Indena SpA E1A17 Trifarma E1A21 LLC Velpharma E1A25 Anuh Pharma E1A31 Chemclone Industries E1A51 Hetero Labs Limited E1B09 Concord Biotech Limited E1B10 ScinoPharm Taiwan Ltd E1B11 Dongkook Pharmaceutical Co., Ltd. E1B19 Shenzhen Salubris Pharmaceuticals Co., Ltd E1B22 GfM mbH E1B25 Leawell International Ltd E1B28 DCS Pharma AG E1B31 Agno Pharma E1B32 Newchem Spa E1B35 APEX HEALTHCARE LIMITED E1B51 AMRI E1C21 Aarti Drugs Limited E1C25 Espee Group Innovators E1C31 Ruland Chemical Co., Ltd. E1C32 Merck Chemicals (Shanghai) Co., Ltd. E1C51 Mediking Pharmaceutical Group Ltd E1C57 珠海联邦制药股份有限公司/The United E1D01 Laboratories International Holdings Ltd. FMC Corporation E1D02 Kingchem (Liaoning) Chemical Co., Ltd E1D10 Doosan Corporation E1D22 Sunasia Co., Ltd. E1D25 Bolon Pharmachem Co., Ltd. E1D26 Savior Lifetec Corporation E1D27 Alchem International Pvt Ltd E1D31 Polish Investment and Trade Agency E1D57 Fischer Chemicals AG E1E01 NGL Fine Chem Limited E1E24 常州艾柯轧辊有限公司/ECCO Roller E1E25 Linnea SA E1E26 Everlight Chemical Industrial Corporation E1E27 HARMAN FINOCHEM E1E28 Zhechem Co Ltd E1F01 Midas Pharma GmbH Shanghai Representativ E1F03 Supriya Lifescience Ltd E1F10 KOA Shoji Co Ltd E1F22 NOF Corporation E1F24 上海贺利氏工业技术材料有限公司/Heraeus E1F26 Materials Technology Shanghai Ltd. Novacyl Asia Pacific Ltd E1F28 PharmSol Europe Limited E1F32 Bachem AG E1F35 Louston International Inc. E1F51 High Science Co Ltd E1F55 Chemsphere Technology Inc. E1F57a PharmaCore Biotech Co., Ltd. E1F57b Rockwood Lithium GmbH E1G51 Sarv Bio Labs Pvt Ltd E1G57 抗病毒类、抗肿瘤类、抗感染类和甾体类中间体、原料药和药物制剂及医药合约研发和加工服务 上海创诺医药集团有限公司/Shanghai Desano APIs and Finished products of ARV, Oncology, Anti-infection and Hormone drugs and E1H01 Pharmaceuticals Co., Ltd. -

The Effects of Evodiamine on Serum Total Cholesterol
Arch Biol Sci. 2016;68(3):561-566 DOI:10.2298/ABS150904046Y THE EFFECTS OF EVODIAMINE ON SERUM TOTAL CHOLESTEROL AND TRIGLYCERIDE LEVELS ARE ASSOCIATED WITH THE ACTIVATION OF THE AMPK SIGNALING PATHWAY IN RATS WITH HYPERLIPEMIA Hui Yu1, Hai Hu2, Wuzhuang Gong3, Li Yang3, Zhanli Wang3,4,* and Cuifeng Wang3,* 1 The Second Affiliated Hospital, Baotou Medical College, Baotou 014030, China 2 Department of Pathophysiology, Baotou Medical College, Baotou 014060, China 3 The First Affiliated Hospital, Baotou Medical College, Baotou 014010, China 4 School of Public Health, Baotou Medical College, Baotou 014060, China *Corresponding authors: [email protected]; [email protected] Received: September 4, 2015; Revised: October 14, 2015; Accepted: October 26, 2015; Published online: May 9, 2016 Abstract: Evodiamine, a naturally occurring indole alkaloid, has been reported to have numerous biological activities, including antitumor, antimicrobial and anti-inflammatory effects. Previous studies also suggest that evodiamine prevents obesity. In this study, we confirmed that evodiamine lowered the levels of serum total cholesterol (TC) and triglycerides (TG) in rats with hyperlipemia. Furthermore, our findings suggest that the activation of the AMP-activated protein kinase (AMPK) pathway might contribute in part to the effect of evodiamine on the serum levels of TC and TG. Key words: evodiamine; AMP-activated protein kinase; total cholesterol; triglycerides INTRODUCTION evodiamine inhibited neuropeptide Y (NPY) mRNA and peptide levels in the arcuate nucleus (ARC) of Evodiamine is a bioactive compound present in the the hypothalamus, which might be one of the mecha- fruit of Evodia rutaecarpa (Juss.) Benth [1]. Initially, it nisms by which evodiamine exerted its anti-obesity was identified as a vanilloid receptor 1 (TRPV1) ago- effects. -

Transient Receptor Potential Channels As Drug Targets: from the Science of Basic Research to the Art of Medicine
1521-0081/66/3/676–814$25.00 http://dx.doi.org/10.1124/pr.113.008268 PHARMACOLOGICAL REVIEWS Pharmacol Rev 66:676–814, July 2014 Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics ASSOCIATE EDITOR: DAVID R. SIBLEY Transient Receptor Potential Channels as Drug Targets: From the Science of Basic Research to the Art of Medicine Bernd Nilius and Arpad Szallasi KU Leuven, Department of Cellular and Molecular Medicine, Laboratory of Ion Channel Research, Campus Gasthuisberg, Leuven, Belgium (B.N.); and Department of Pathology, Monmouth Medical Center, Long Branch, New Jersey (A.S.) Abstract. ....................................................................................679 I. Transient Receptor Potential Channels: A Brief Introduction . ...............................679 A. Canonical Transient Receptor Potential Subfamily . .....................................682 B. Vanilloid Transient Receptor Potential Subfamily . .....................................686 C. Melastatin Transient Receptor Potential Subfamily . .....................................696 Downloaded from D. Ankyrin Transient Receptor Potential Subfamily .........................................700 E. Mucolipin Transient Receptor Potential Subfamily . .....................................702 F. Polycystic Transient Receptor Potential Subfamily . .....................................703 II. Transient Receptor Potential Channels: Hereditary Diseases (Transient Receptor Potential Channelopathies). ......................................................704 -

Preclinical and Clinical Evidence of Therapeutic Agents for Paclitaxel-Induced Peripheral Neuropathy
International Journal of Molecular Sciences Review Preclinical and Clinical Evidence of Therapeutic Agents for Paclitaxel-Induced Peripheral Neuropathy Takehiro Kawashiri 1,* , Mizuki Inoue 1, Kohei Mori 1, Daisuke Kobayashi 1, Keisuke Mine 1, Soichiro Ushio 2, Hibiki Kudamatsu 1, Mayako Uchida 3 , Nobuaki Egashira 4 and Takao Shimazoe 1 1 Department of Clinical Pharmacy and Pharmaceutical Care, Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka 812-8582, Japan; [email protected] (M.I.); [email protected] (K.M.); [email protected] (D.K.); [email protected] (K.M.); [email protected] (H.K.); [email protected] (T.S.) 2 Department of Pharmacy, Okayama University Hospital, Okayama 700-8558, Japan; [email protected] 3 Department of Clinical Pharmacy, Faculty of Pharmaceutical Sciences, Doshisha Women’s College of Liberal Arts, Kyoto 610-0395, Japan; [email protected] 4 Department of Pharmacy, Kyushu University Hospital, Fukuoka 812-8582, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-92-642-6573 Abstract: Paclitaxel is an essential drug in the chemotherapy of ovarian, non-small cell lung, breast, gastric, endometrial, and pancreatic cancers. However, it frequently causes peripheral neuropathy as a dose-limiting factor. Animal models of paclitaxel-induced peripheral neuropathy (PIPN) have been established. The mechanisms of PIPN development have been elucidated, and many drugs and agents have been proven to have neuroprotective effects in basic studies. -

Neuroprotective Studies of Evodiamine in an Okadaic Acid-Induced Neurotoxicity
Article Neuroprotective Studies of Evodiamine in an Okadaic Acid-Induced Neurotoxicity Ching-Hsuan Chou and Chia-Ron Yang * School of Pharmacy, College of Medicine, National Taiwan University, Taipei 10050, Taiwan; [email protected] * Correspondence: [email protected]; Tel.: +886-2-3366-8758 Abstract: Background: Alzheimer’s disease (AD) is the most common neurodegenerative disease, and it manifests as progressive memory loss and cognitive decline. However, there are no effective therapies for AD, which is an urgent problem to solve. Evodiamine, one of the main bioactive ingredi- ents of Evodia rutaecarpa, has been reported to ameliorate blood–brain barrier (BBB) permeability and improve cognitive impairment in ischemia and AD mouse models. However, whether evodiamine alleviates tauopathy remains unclear. This study aimed to examine whether evodiamine ameliorates tau phosphorylation and cognitive deficits in AD models. Methods: A protein phosphatase 2A inhibitor, okadaic acid (OA), was used to induce tau phosphorylation to mimic AD-like models in neuronal cells. Protein expression and cell apoptosis were detected using Western blotting and flow cytometry, respectively. Spatial memory/cognition was assessed using water maze, passive avoidance tests, and magnetic resonance imaging assay in OA-induced mice models, and brain slices were evaluated further by immunohistochemistry. Results: The results showed that evodiamine significantly reduced the expression of phosphor-tau, and further decreased tau aggregation and neuronal cell death in response to OA treatment. This inhibition was found to be via the inhibition Citation: Chou, C.-H.; Yang, C.-R. of glycogen synthase kinase 3β, cyclin-dependent kinase 5, and mitogen-activated protein kinase Neuroprotective Studies of pathways. -

Dr. Duke's Phytochemical and Ethnobotanical Databases List of Chemicals for Epilepsy
Dr. Duke's Phytochemical and Ethnobotanical Databases List of Chemicals for Epilepsy Chemical Activity Count (+)-ALPHA-VINIFERIN 1 (+)-BORNYL-ISOVALERATE 1 (+)-CATECHIN 3 (+)-EUDESMA-4(14),7(11)-DIENE-3-ONE 1 (+)-HERNANDEZINE 1 (+)-ISOCORYDINE 1 (+)-PSEUDOEPHEDRINE 1 (+)-SYRINGARESINOL-DI-O-BETA-D-GLUCOSIDE 1 (+)-T-CADINOL 1 (-)-16,17-DIHYDROXY-16BETA-KAURAN-19-OIC 1 (-)-ALPHA-BISABOLOL 2 (-)-ANABASINE 1 (-)-APOGLAZIOVINE 1 (-)-BETONICINE 1 (-)-BORNYL-CAFFEATE 1 (-)-BORNYL-FERULATE 1 (-)-BORNYL-P-COUMARATE 1 (-)-DICENTRINE 2 (-)-EPIAFZELECHIN 1 (-)-EPICATECHIN 1 (-)-EPIGALLOCATECHIN-GALLATE 1 (1'S)-1'-ACETOXYCHAVICOL-ACETATE 1 (15:1)-CARDANOL 1 (E)-4-(3',4'-DIMETHOXYPHENYL)-BUT-3-EN-OL 1 1,7-BIS-(4-HYDROXYPHENYL)-1,4,6-HEPTATRIEN-3-ONE 1 1,8-CINEOLE 4 10-ACETOXY-8-HYDROXY-9-ISOBUTYLOXY-6-METHOXYTHYMOL 1 Chemical Activity Count 10-DEHYDROGINGERDIONE 1 10-GINGERDIONE 1 11-HYDROXY-DELTA-8-THC 1 11-HYDROXY-DELTA-9-THC 1 13',II8-BIAPIGENIN 1 13-OXYINGENOL-ESTER 1 16,17-DIHYDROXY-16BETA-KAURAN-19-OIC 1 16-EPIMETHUENINE 1 16-HYDROXYINGENOL-ESTER 1 2'-O-GLYCOSYLVITEXIN 1 2-BETA,3BETA-27-TRIHYDROXYOLEAN-12-ENE-23,28-DICARBOXYLIC-ACID 1 2-METHYLBUT-3-ENE-2-OL 2 20-DEOXYINGENOL-ESTER 1 22BETA-ESCIN 1 24-METHYLENE-CYCLOARTANOL 1 3,3'-DIMETHYLELLAGIC-ACID 1 3,4-DIMETHOXYTOLUENE 1 3,4-METHYLENE-DIOXYCINNAMIC-ACID-BORNYL-ESTER 2 3,4-SECOTRITERPENE-ACID-20-EPI-KOETJAPIC-ACID 1 3-ACETYLACONITINE 1 3-ACETYLNERBOWDINE 1 3-BETA-HYDROXY-2,3-DIHYDROWITHANOLIDE-F 1 3-HYDROXY-FLAVONE 1 3-N-BUTYL-PHTHALIDE 3 3-O-ACETYLOLEANOLIC-ACID 1 3-OXO-11-ALPHA-HYDROXYOLEAN-12-ENE-30-OIC-ACID -

A 1A9, A431 and A549 Cell Lines, 3478 12Α,13Α-Aziridinyl Epothilone
Index A Acanthoscelides obtectus, 4091 1A9, A431 and A549 cell lines, 3478 Acanthostrongylophora, 1293 12a,13a-Aziridinyl epothilone derivatives, 990 Acari, 4070, 4083 Aaptos ciliata, 1292 Acaricides, 4070, 4076, 4083 AATs. See Anthocyanin acyltransferases Accelerated solvent extraction (ASE), 1017, (AATs) 2017, 2021–2023, 2032, 2037, 2111 Ab (1-42), 2287 p-Acceptors, 692 ABA biosynthesis, 1736 Accretion, 2405 Ab aggregation, 2286 Accumulation, 2321 Abdominal troubles, 3477 Acetaldehyde (ATL), 2902 Aberrant behavior, 1375 Acetaldehyde acid, 1783 Aberrant estrous cycles, 2421 Acetate, 50, 59 Abies alba, 2980 Acetate-mevalonate (Ac-MEV), 2943 Abietadiene synthase, 2725 Acetate pathway, 2314 Abiotic and biotic elicitors, 2783 Acetic acid (ACE), 851, 1608, 2884 Abiotic factor, 1697 Acetogenic bacteria, 2443 Abiotic stresses, 2930 Acetone, 3371 Ab neuropathology, 2285 Acetonitrile, 691, 1170, 1175 Ab oligomerization, 2285 3-Acetylaconitine, 1508 Ab oligomers, 2283 Acetyl-ACP, 963 Ab peptides, 1308, 2285 Acetyl-and butyrylcholinesterase inhibitors, Ab polymerization, 2287 3488 Abrin, 296 Acetylcholine (ACh), 1241, 1306, 1333, 1526, Abscisic acid (ABA), 2860, 3591 1527, 1529, 1530, 3710 Absolute configuration, 930, 3320 Acetylcholine esterase, 51, 52 Absolute configuration of monosaccharides, Acetylcholine esterase inhibitor activity, 2906 3319 Acetylcholinesterase (AChE), 41, 1246, 1308, Absorbance (A), 3334, 3380 1334, 1527, 1529–1533, 1535, 4097 Absorbance spectrum, 3929, 3933, 3934 Acetyl-CoA, 61, 584 Absorption, 1230, 2321, 2470–2476, Acetyl-CoA carboxylase (ACC), 1658, 1825 2478–2481, 3134, 3377, 3616, 4024 3-Acetyldeoxynivalenol, 3127, 3145 coefficient, 3380 15-Acetyl deoxynivalenol (15ADON), and metabolism, 1428 3127, 3145 wavelength, 4028 Acetylerucifoline, 1061 Abusive correlations, 2410 Acetylgaertneroside, 3054 Acacetin, 1830 60-O-Acetylgeniposide, 3058 Acacia, 865 8-O-Acetylharpagide, 3055 a2C adrenergic, 4124 Acetylintermedine, 1061 Acanthaceae, 801 Acetyllycopsamine, 1061 K.G. -

Supplement Ii to the Japanese Pharmacopoeia Seventeenth Edition
SUPPLEMENT II TO THE JAPANESE PHARMACOPOEIA SEVENTEENTH EDITION O‹cial from June 28, 2019 English Version THE MINISTRY OF HEALTH, LABOUR AND WELFARE Notice: This English Version of the Japanese Pharmacopoeia is published for the convenience of users unfamiliar with the Japanese language. When and if any discrepancy arises between the Japanese original and its English translation, the former is authentic. Printed in Japan The Ministry of Health, Labour and Welfare Ministerial Notification No. 49 Pursuant to Paragraph 1, Article 41 of Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices (Act No. 145, 1960), this notification stated that a part of the Japanese Pharmacopoeia was revised as follows*. NEMOTO Takumi The Minister of Health, Labour and Welfare June 28, 2019 A part of the Japanese Pharmacopoeia (Ministerial Notification No. 64, 2016) was revised as follows*. (The text referred to by the term ``as follows'' are omitted here. All of the revised Japanese Pharmacopoeia in accordance with this notification (hereinafter referred to as ``new Pharmacopoeia'' in Supplement 2) are made available for public exhibition at the Pharmaceutical Evaluation Division, Pharmaceutical Safety and Environmen- tal Health Bureau, Ministry of Health, Labour and Welfare, at each Regional Bureau of Health and Welfare, and at each Prefectural Office in Japan). Supplementary Provisions (Effective Date) Article 1 This Notification is applied from June 28, 2019. (Transitional measures) Article 2 In the case of drugs which are listed in the Japanese Pharmacopoeia (hereinafter referred to as ``previous Pharmacopoeia'') [limited to those listed in new Pharmacopoeia] and drugs which have been approved as of June 28, 2019 as prescribed under Paragraph 1, Article 14 of Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices [including drugs the Minister of Health, Labour and Welfare specifies (the Ministry of Health and Welfare Ministerial Notification No. -

UNIVERSITY of CAPE TOWN for a Dissertation in Partial Fulfilment of the Requirements for the Degree Town Mphil (Biomedical Forensic Science)
Complementary and Alternate Medicines: A Forensic Analysis of the Potential Adulteration of Over-The-Counter Anorectics and “Lifestyle” Medicines in South Africa By Sandra Lynne Catterson CTTSAN003 SUBMITTED TO THE UNIVERSITY OF CAPE TOWN For a dissertation in partial fulfilment of the requirements for the degree Town MPhil (Biomedical Forensic Science) Faculty of HealthCape Sciences UNIVERSITY ofOF CAPE TOWN 14th August 2017 Division of Forensic Medicine and Toxicology Department of Pathology University of CapeUniversity Town Supervisor: Bronwen Davies; Division of Forensic Medicine and Toxicology Co-supervisors: Professor Peter Smith; Division of Pharmacology Marie Kathrina Mendoza Aukloo; Division of Forensic Medicine and Toxicology i The copyright of this thesis vests in the author. No quotation from it or information derived from it is to be published without full acknowledgementTown of the source. The thesis is to be used for private study or non- commercial research purposes only. Cape Published by the University ofof Cape Town (UCT) in terms of the non-exclusive license granted to UCT by the author. University DECLARATION I, Sandra Catterson, hereby declare that the work on which this dissertation is based is my original work (except where acknowledgements indicate otherwise) and that neither the whole work nor any part of it has been, is being, or is to be submitted for another degree in this or any other university. I empower the university to reproduce for the purpose of research either the whole or any portion of the content in any manner whatsoever. 14th August 2017 Signature removed i ABSTRACT Background: Complementary and Alternate Medicines (CAMs) in South Africa are not yet subjected to the same rigorous testing required for allopathic (prescription) medication, yet they are freely available as over-the-counter medicines.