Conditional Activation of NRG1 Signaling in the Brain Modulates Cortical Circuitry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of LIM Kinase 1 and Its Substrates in Cell Cycle Progression
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2014 The Role of LIM Kinase 1 and its Substrates in Cell Cycle Progression Lisa Ritchey University of Central Florida Part of the Medical Sciences Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Doctoral Dissertation (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Ritchey, Lisa, "The Role of LIM Kinase 1 and its Substrates in Cell Cycle Progression" (2014). Electronic Theses and Dissertations, 2004-2019. 1300. https://stars.library.ucf.edu/etd/1300 THE ROLE OF LIM KINASE 1 AND ITS SUBSTRATES IN CELL CYCLE PROGRESSION by LISA RITCHEY B.S. Florida State University 2007 M.S. University of Central Florida 2010 A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Burnett School of Biomedical Sciences in the College of Graduate Studies at the University of Central Florida Orlando, Florida Summer Term 2014 Major Professor: Ratna Chakrabarti © 2014 Lisa Ritchey ii ABSTRACT LIM Kinase 1 (LIMK1), a modulator of actin and microtubule dynamics, has been shown to be involved in cell cycle progression. In this study we examine the role of LIMK1 in G1 phase and mitosis. We found ectopic expression of LIMK1 resulted in altered expression of p27Kip1, the G1 phase Cyclin D1/Cdk4 inhibitor. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Deep Learning–Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer Kumardeep Chaudhary1, Olivier B
Published OnlineFirst October 5, 2017; DOI: 10.1158/1078-0432.CCR-17-0853 Statistics in CCR Clinical Cancer Research Deep Learning–Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer Kumardeep Chaudhary1, Olivier B. Poirion1, Liangqun Lu1,2, and Lana X. Garmire1,2 Abstract Identifying robust survival subgroups of hepatocellular car- index (C-index) ¼ 0.68]. More aggressive subtype is associated cinoma (HCC) will significantly improve patient care. Current- with frequent TP53 inactivation mutations, higher expression ly, endeavor of integrating multi-omicsdatatoexplicitlypredict of stemness markers (KRT19 and EPCAM)andtumormarker HCC survival from multiple patient cohorts is lacking. To fill BIRC5, and activated Wnt and Akt signaling pathways. We this gap, we present a deep learning (DL)–based model on HCC validated this multi-omics model on five external datasets of that robustly differentiates survival subpopulations of patients various omics types: LIRI-JP cohort (n ¼ 230, C-index ¼ 0.75), in six cohorts. We built the DL-based, survival-sensitive model NCI cohort (n ¼ 221, C-index ¼ 0.67), Chinese cohort (n ¼ on 360 HCC patients' data using RNA sequencing (RNA-Seq), 166, C-index ¼ 0.69), E-TABM-36 cohort (n ¼ 40, C-index ¼ miRNA sequencing (miRNA-Seq), and methylation data from 0.77), and Hawaiian cohort (n ¼ 27, C-index ¼ 0.82). This TheCancerGenomeAtlas(TCGA),whichpredictsprognosis is the first study to employ DL to identify multi-omics features as good as an alternative model where genomics and clinical linked to the differential survival of patients with HCC. Given data are both considered. This DL-based model provides two its robustness over multiple cohorts, we expect this workflow to optimal subgroups of patients with significant survival differ- be useful at predicting HCC prognosis prediction. -

Characterization of Gf a Drosophila Trimeric G Protein Alpha Subunit
Characterization of Gf a Drosophila trimeric G protein alpha subunit Naureen Quibria Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2012 © 2012 Naureen Quibria All rights reserved Abstract Characterization of Gf a Drosophila trimeric G-protein alpha subunit Naureen Quibria In the morphogenesis of tissue development, how coordination of patterning and growth achieve the correct organ size and shape is a principal question in biology. Efficient orchestrating mechanisms are required to achieve this and cells have developed sophisticated systems for reception and interpretation of the multitude of extracellular stimuli to which they are exposed. Plasma membrane receptors play a key role in the transmission of such signals. G-protein coupled receptors (GPCRs) are the largest class of cell surface receptors that respond to an enormous diversity of extracellular stimuli, and are critical mediators of cellular signal transduction in eukaryotic organisms. Signaling through GPCRs has been well characterized in many biological contexts. While they are a major class of signal transducers, there are not many defined instances where GPCRs have been implicated in the process of development to date. The Drosophila wing provides an ideal model system to elucidate and address the role of GPCRs in development, as its growth is regulated by a small number of conserved signaling pathways. In my thesis work, I address the role of a trimeric G alpha protein in Drosophila, Gαf, and what part it may play in development. In particular, I explore the role of Gαf as an alpha subunit of a trimeric complex, to determine what heptahelical receptors might act as its cognate receptor. -

PI3K Catalytic Isoform Alteration Promotes the LIMK1-Related
ANTICANCER RESEARCH 37 : 1805-1818 (2017) doi:10.21873/anticanres.11515 PI3K Catalytic Isoform Alteration Promotes the LIMK1-related Metastasis Through the PAK1 or ROCK1/2 Activation in Cigarette Smoke-exposed Ovarian Cancer Cells GA BIN PARK 1 and DAEJIN KIM 2 1Department of Biochemistry, Kosin University College of Medicine, Busan, Republic of Korea; 2Department of Anatomy, Inje University College of Medicine, Busan, Republic of Korea Abstract. Aim: To investigate the molecular mechanisms Several studies have shown a strong correlation between by which long-term exposure to cigarette smoke extract cigarette smoke (CS) and cancer metastasis through the (CSE) contributes to ovarian cancer metastasis. Materials induction of numerous factors involved in migration activity and Methods: Western blot analysis for diverse p110 (1-3). The exposure to CS induces the epithelial- isoforms of phosphoinositide 3-kinase (PI3K)-related mesenchymal transition (EMT) process and up-regulates the signaling pathway and epithelial-mesenchymal transition expression of EMT markers, including N-cadherin and (EMT) markers was performed to analyze the underlying vimentin (4, 5). Cigarette smoke extract (CSE) treatment mechanisms. Migratory activity of CSE-exposed ovarian significantly induces interleukin-8 (IL-8) and transforming cancer cells was determined by transendothelial migration growth factor-beta 1 (TGF- β1 ) production and profoundly and invasion assay. Results: After exposure to CSE for four suppresses the proliferation and growth of erythroid and weeks, CaOV3 (primary) and SKOV3 (metastatic) ovarian granulocyte-macrophage progenitors (6). Stimulation with cancer cells showed enhanced mesenchymal characteristics CSE in human lung fibroblast cells induces the expression and produced EMT-related cytokines [intwerleukin-8 (IL-8), of phosphorylated Smad3, a main downstream target of the vascular endothelial growth factor (VEGF) and TGF- β1 receptor, which results in the secretion of vascular transforming growth factor-beta 1 (TGF- β1 )]. -

Gene Networks Activated by Specific Patterns of Action Potentials in Dorsal Root Ganglia Neurons Received: 10 August 2016 Philip R
www.nature.com/scientificreports OPEN Gene networks activated by specific patterns of action potentials in dorsal root ganglia neurons Received: 10 August 2016 Philip R. Lee1,*, Jonathan E. Cohen1,*, Dumitru A. Iacobas2,3, Sanda Iacobas2 & Accepted: 23 January 2017 R. Douglas Fields1 Published: 03 March 2017 Gene regulatory networks underlie the long-term changes in cell specification, growth of synaptic connections, and adaptation that occur throughout neonatal and postnatal life. Here we show that the transcriptional response in neurons is exquisitely sensitive to the temporal nature of action potential firing patterns. Neurons were electrically stimulated with the same number of action potentials, but with different inter-burst intervals. We found that these subtle alterations in the timing of action potential firing differentially regulates hundreds of genes, across many functional categories, through the activation or repression of distinct transcriptional networks. Our results demonstrate that the transcriptional response in neurons to environmental stimuli, coded in the pattern of action potential firing, can be very sensitive to the temporal nature of action potential delivery rather than the intensity of stimulation or the total number of action potentials delivered. These data identify temporal kinetics of action potential firing as critical components regulating intracellular signalling pathways and gene expression in neurons to extracellular cues during early development and throughout life. Adaptation in the nervous system in response to external stimuli requires synthesis of new gene products in order to elicit long lasting changes in processes such as development, response to injury, learning, and memory1. Information in the environment is coded in the pattern of action-potential firing, therefore gene transcription must be regulated by the pattern of neuronal firing. -

Functional Analysis of the LIM Kinase 1 and Its Role in Cell Cycle Progression
Functional Analysis of the LIM kinase 1 and its role in Cell Cycle progression Inauguraldissertation zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Ayça Sayı aus Istanbul (Türkei) Zürich 2008 Genehmigt von der Philosophisch- Naturwissenschaftlichen Fakultät auf Antrag von Prof. Dr. Wilhelm Krek, Prof.Dr. Matthias Peter und Prof. Dr. Denis Monard. Basel, den 20. Juni 2006 Prof. Dr. Hans- Jakob Wirz Dekan Abstract LIMK1 (LIM-kinase 1) is a member of the LIMKs family of serine/threonine kinases, comprised of LIMK1, LIMK2 and testicular specific kinases, TESK1 and TESK2. These enzymes catalyze phosphorylation of an actin depolymerizing factor cofilin, thereby inactivate its depolymerizing activity, that leads to actin stabilization as filamentous actin (F-actin). As a consequence, LIMK1 plays a key role in actin cytoskeleton remodeling, notably in response to many extracellular cues that trigger the activation of the small Rho GTPases Rho, Rac and Cdc42, these latter being able to activate LIMK1 through their downstream effectors ROCK, PAK1 and PAK4 respectively. Moreover, on a pathological point of view, dysregulation of LIMK1 has being associated to several diseases: indeed, partial loss of LIMK1 is associated to a neurological disorder called William’s syndrome, whereas LIMK1 upregulation and/or hyperactivation is linked to cancer metastasis. The introduction chapter of this thesis details the current knowledge about LIMK1 and its func- tion in cell migration, cell cycle, neuronal differentiation, and phagocytosis. Additionally, the contribution of LIMK1 dysregulation in pathological circumstances, notably in WilliamÕs syn- drome and in cancer metastasis is discussed. The results chapter summarizes the work undertaken as partial fulfilment of this doctoral study. -

Low-Molecular-Weight Fucoidan Induces Endothelial Cell Migration
Low-Molecular-Weight Fucoidan Induces Endothelial Cell Migration via the PI3K/AKT Pathway and Modulates the Transcription of Genes Involved in Angiogenesis Claire Bouvard, Isabelle Galy-Fauroux, Françoise Grelac, Wassila Carpentier, Anna Lokajczyk, Sophie Gandrille, Sylvia Colliec-Jouault, Anne-Marie Fischer, Dominique Helley To cite this version: Claire Bouvard, Isabelle Galy-Fauroux, Françoise Grelac, Wassila Carpentier, Anna Lokajczyk, et al.. Low-Molecular-Weight Fucoidan Induces Endothelial Cell Migration via the PI3K/AKT Pathway and Modulates the Transcription of Genes Involved in Angiogenesis. Marine drugs, MDPI, 2015, 13 (12), pp.7446 - 7462. 10.3390/md13127075. hal-01514447 HAL Id: hal-01514447 https://hal.archives-ouvertes.fr/hal-01514447 Submitted on 3 May 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Article Low-Molecular-Weight Fucoidan Induces Endothelial Cell Migration via the PI3K/AKT Pathway and Modulates the Transcription of Genes Involved in Angiogenesis -

Promoter Methylation Profiles Between Human Lung
1310 Regular Article Biol. Pharm. Bull. 36(8) 1310–1316 (2013) Vol. 36, No. 8 Promoter Methylation Profiles between Human Lung Adenocarcinoma Multidrug Resistant A549/Cisplatin (A549/DDP) Cells and Its Progenitor A549 Cells Ruiling Guo,*,a Guoming Wu,b,# Haidong Li,a Pin Qian,c Juan Han,d Feng Pan,a Wenbi Li,a Jin Li,b and Fuyun Ji*,b a Department of Respiratory Diseases, 324th Hospital of the People’s Liberation Army; d Department of Emergency Medicine, 324th Hospital of People’s Liberation Army; Chongqing 400020, China: b Institute of Human Respiratory Diseases, Xinqiao Hospital, Third Military Medical University; and c Institute of Field Internal Medicine, Xinqiao Hospital, Third Military Medical University; Chongqing 400037, China. Received February 19, 2013; accepted May 27, 2013 Although aberrant DNA methylation has been implicated in the pathophysiology of lung cancer, the role of methylation in multidrug resistance (MDR) of lung cancer has remained unclear. To investigate whether certain distinct DNA methylation pattern is associated with acquired MDR of lung adenocarcinoma, meth- ylated-DNA immunoprecipitation-chromatin immunoprecipitation (MeDIP-ChIP) was utilised to compare the genome-wide promoter methylation of the human lung adenocarcinoma MDR A549/cisplatin (A549/DDP) cells with its progenitor A549 cells. The comparison identified 3617 genes with differentially methylated promoter, of which 1581 were hypermethylated and 2036 were hypomethylated. Then, bisulphite sequenc- ing polymerase chain reaction (PCR) (BSP) and quantitative reverse transcription (RT)-PCR (Q-PCR) were used to validate the promoter methylation of five candidate genes and to determine whether the expression of genes was associated with the promoter methylation. -

Itga4 Cldn16 Cldn9 Cldn15 Cldn22 Ocln Esam
Supplementary material J Med Genet Table S1. List of 263 genes included in the AGS-LEUK panel. Axonal Guidance Signaling genes as AGS and Leukocyte transvasation genes as LEUK. List of genes (AGS) List of genes (LEUK) ABLIM1 CLDN11 ACTR3 MMP14 ADAM11 MMP15 ADAM23 CTNNA1 ADAMTS1 ENSG00000130396 ADAMTS4 CLDN6 ADAMTS9 MMP24 ARHGEF15 ARHGAP12 ARHGEF6 DLC1 ARPC1B TIMP2 BDNF RAPGEF3 BMP1 F11R BMP4 CLDN23 BMP6 CLDN8 BMP7 JAM3 CXCL12 CLDN3 CXCR4 ARHGAP8 DPYSL5 ICAM1 EFNA1 MMP16 EFNA5 JAM2 ENPEP CLDN7 EPHA1 TIMP3 EPHA3 VCAM1 EPHA5 CLDN5 EPHA7 MSN EPHB1 NOX3 EPHB2 ACTC1 EPHB4 VAV2 FGFR2 CLDN10 FZD1 RAP1GAP FZD10 VAV3 FZD5 MAPK10 FZD6 CTNNA2 GAB1 CDH5 GLI1 PECAM1 GLI3 CTNND1 GNA14 ITGA4 GNAI1 CLDN16 GNAO1 CLDN9 GNAS CLDN15 GNB4 CLDN22 GNG11 OCLN GNG2 ESAM Gallego-Martinez A, et al. J Med Genet 2019; 0:1–7. doi: 10.1136/jmedgenet-2019-106159 Supplementary material J Med Genet GNG7 ACTB IGF1 CYBA IRS1 CTNNB1 IRS2 MMP9 ITGA3 MAPK14 ITGB1 MAPK11 LIMK1 MAPK12 LIMK2 MAPK13 LINGO1 PRKCB LRRC4C PXN MME BCAR1 MMP11 THY1 MMP2 ARHGAP5 MRAS MYL2 MYL9 MYLPF NFATC4 RAP1A NGFR RAP1B NOTUM VASP NRP1 ACTN4 NTN3 ACTN1 NTRK2 VCL NTRK3 RAPGEF4 PAK3 ITK PAK4 VAV1 PAPPA2 PDGFA PDGFC PIK3CB PIK3R1 PLCE1 PLCH1 PLCH2 PLXNA2 PLXNB1 PLXND1 PPP3CA PRKACB PRKAR2A PRKAR2B PRKCA PRKCZ PRKD3 ROBO2 SDC2 SDCBP Gallego-Martinez A, et al. J Med Genet 2019; 0:1–7. doi: 10.1136/jmedgenet-2019-106159 Supplementary material J Med Genet SEMA3B SEMA3C SEMA3E SEMA3F SEMA4F SEMA4G SEMA5A SEMA6B SEMA6D SEMA7A SHC1 SLIT2 SLIT3 STK36 TUBA4A TUBB2B TUBB4A TUBB4B TUBB6 UNC5C UNC5D ENSG00000165197 WIPF1 WNT3 WNT5A WNT7A WNT7B NTNG1 NTNG2 LRRC4 NTN4 TRPC1 TRPC3 TRPC6 PPP3CB PPP3CC PPP3R1 NFATC2 NFATC3 PTK2 FYN RAC1 CDC42 ABLIM2 NCK1 PAK1 PAK2 Gallego-Martinez A, et al. -

Calcineurin Is an Important Factor Involved in Glucose Uptake in Human Adipocytes
Molecular and Cellular Biochemistry (2018) 445:157–168 https://doi.org/10.1007/s11010-017-3261-0 Calcineurin is an important factor involved in glucose uptake in human adipocytes Ana Catarina R. G. Fonseca1,2 · Eugénia Carvalho2,3,4,5 · Jan W. Eriksson1 · Maria J. Pereira1 Received: 27 October 2017 / Accepted: 23 December 2017 / Published online: 27 January 2018 © The Author(s) 2018. This article is an open access publication Abstract Calcineurin inhibitors are used in immunosuppressive therapy applied after transplantation, but they are associated with major metabolic side effects including the development of new onset diabetes. Previously, we have shown that the calcineu- rin inhibiting drugs tacrolimus and cyclosporin A reduce adipocyte and myocyte glucose uptakes by reducing the amount of glucose transporter type 4 (GLUT4) at the cell surface, due to an increased internalization rate. However, this happens without alteration in total protein and phosphorylation levels of key proteins involved in insulin signalling or in the total amount of GLUT4. The present study evaluates possible pathways involved in the altered internalization of GLUT4 and consequent reduction of glucose uptake provoked by calcineurin inhibitors in human subcutaneous adipose tissue. Short- and long-term treatments with tacrolimus, cyclosporin A or another CNI deltamethrin (herbicide) decreased basal and insulin- dependent glucose uptake in adipocytes, without any additive effects observed when added together. However, no tacrolimus effects were observed on glucose uptake when gene transcription and protein translation were inhibited. Investigation of genes potentially involved in GLUT4 trafficking showed only a small effect on ARHGEF11 gene expression (p < 0.05). In conlusion, the specific inhibition of calcineurin, but not that of protein phosphatases, decreases glucose uptake in human subcutaneous adipocytes, suggesting that calcineurin is an important regulator of glucose transport. -
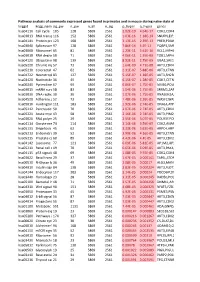
Pathway Analysis of Commonly Expressed Genes Found in Primates and in Mouse During Naïve State of Pluripotenc Keggid Kegg Names
Pathway analysis of commonly expressed genes found in primates and in mouse during naïve state of pluripotency. keggid kegg_namesig_pw n_pw n_all n_sig p_hyper q_hyper genes hsa04110 Cell cycle 105 128 5869 2561 2.02E-19 4.64E-17 CDK2,CDK4,CDK6,CDK7,CDKN1A,CDKN1B,STAG1,CDKN2B,ANAPC10,MAD2L2,STAG2,GADD45G,DBF4,YWHAQ,CHEK1,CHEK2,CREBBP,GADD45A,E2F1,E2F3,E2F4,E2F5,EP300,ORC3,CDC26,ABL1,ANAPC13,SFN,GSK3B,ANAPC2,ANAPC4,HDAC1,HDAC2,MAD2L1,SMAD2,SMAD3,SMAD4,MCM2,MCM3,MCM4,MCM5,MCM6,MCM7,MDM2,MYC,GADD45B,ATM,ORC1,ORC2,ORC4,ORC5,FZR1,ANAPC7,ANAPC11,PLK1,ATR,PRKDC,RAD21,RB1,RBL1,CCND1,ANAPC1,SKP2,BUB1,BUB1B,TFDP1,TFDP2,TGFB1,TGFB2,TTK,WEE1,YWHAB,YWHAE,YWHAG,YWHAH,YWHAZ,ZBTB17,SMC1A,CDC7,CDC45,MAD1L1,CDC14A,CDC23,CDC16,CCNA2,CCNB1,CCND2,CCND3,CCNE1,CCNH,PKMYT1,SMC3,CCNB2,CCNE2,BUB3,PTTG1,ESPL1,CDK1,CDC6,CDC20,CDC25A,CDC25B,CDC25C,CDC27,RBX1 hsa03013 RNA transport116 152 5869 2561 1.03E-16 1.18E-14 SNUPN,EIF1,POP7,SRRM1,SAP18,EIF1B,PRMT5,TACC3,NXF1,RPP30,RPP38,PAIP1,POP4,RPP40,RNPS1,POP1,STRAP,DDX20,XPOT,CLNS1A,NUP35,RPP25L,EEF1A1,EEF1A2,EIF2S1,EIF2B1,EIF4A1,EIF4B,EIF4E,EIF4EBP1,EIF4G1,EIF4G2,EIF5,CASC3,NCBP2,ACIN1,NUP205,NUP210,NUP62,GEMIN5,UPF2,PABPC1,NXT1,NUP43,EIF3E,KPNB1,MAGOH,NCBP1,NUP88,NUP98,GEMIN4,NMD3,POP5,PHAX,NUP54,PNN,RPP25,GEMIN8,MAGOHB,ELAC1,NDC1,NUP133,NXT2,NUP107,THOC2,XPO5,RAN,RANBP2,RANGAP1,SENP2,UPF1,ELAC2,SEC13,UPF3B,SMN1,SUMO3,SUMO2,TPR,SUMO1,XPO1,NUP37,DDX39B,THOC6,GEMIN7,GEMIN6,RPP21,NUP85,THOC7,NUP214,AAAS,SEH1L,THOC3,RAE1,THOC5,EIF3A,EIF3B,EIF3C,EIF3D,EIF3F,EIF3G,EIF3H,EIF3I,EIF4G3,PABPC4,EIF2B4,EIF2B2,EIF2B5,EIF2S2,EIF4E2,NUP155,EIF5B,TGS1,NUP93,EIF4A3,NUP153,THOC1