Analysis of Chromatin-State Plasticity Identifies Cell-Type–Specific Regulators of H3k27me3 Patterns
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Biomarkers of Key Mirnas and Target Genes Associated with Acute Myocardial Infarction
The biomarkers of key miRNAs and target genes associated with acute myocardial infarction Qi Wang1, Bingyan Liu2,3, Yuanyong Wang4, Baochen Bai1, Tao Yu3 and Xian–ming Chu1,5 1 Department of Cardiology, The Affiliated hospital of Qingdao University, Qingdao, China 2 School of Basic Medicine, Qingdao University, Qingdao, China 3 Institute for Translational Medicine, Qingdao University, Qingdao, China 4 Department of Thoracic Surgery, Affiliated Hospital of Qingdao University, Qingdao, China 5 Department of Cardiology, The Affiliated Cardiovascular Hospital of Qingdao University, Qingdao, China ABSTRACT Background. Acute myocardial infarction (AMI) is considered one of the most prominent causes of death from cardiovascular disease worldwide. Knowledge of the molecular mechanisms underlying AMI remains limited. Accurate biomarkers are needed to predict the risk of AMI and would be beneficial for managing the incidence rate. The gold standard for the diagnosis of AMI, the cardiac troponin T (cTnT) assay, requires serial testing, and the timing of measurement with respect to symptoms affects the results. As attractive candidate diagnostic biomarkers in AMI, circulating microRNAs (miRNAs) are easily detectable, generally stable and tissue specific. Methods. The Gene Expression Omnibus (GEO) database was used to compare miRNA expression between AMI and control samples, and the interactions between miRNAs and mRNAs were analysed for expression and function. Furthermore, a protein-protein interaction (PPI) network was constructed. The miRNAs identified in the bioinformatic analysis were verified by RT-qPCR in an H9C2 cell line. The miRNAs in plasma samples from patients with AMI (n D 11) and healthy controls (n D 11) were used to construct Submitted 23 December 2019 receiver operating characteristic (ROC) curves to evaluate the clinical prognostic value Accepted 14 April 2020 of the identified miRNAs. -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -

Table SII. Significantly Differentially Expressed Mrnas of GSE23558 Data Series with the Criteria of Adjusted P<0.05 And
Table SII. Significantly differentially expressed mRNAs of GSE23558 data series with the criteria of adjusted P<0.05 and logFC>1.5. Probe ID Adjusted P-value logFC Gene symbol Gene title A_23_P157793 1.52x10-5 6.91 CA9 carbonic anhydrase 9 A_23_P161698 1.14x10-4 5.86 MMP3 matrix metallopeptidase 3 A_23_P25150 1.49x10-9 5.67 HOXC9 homeobox C9 A_23_P13094 3.26x10-4 5.56 MMP10 matrix metallopeptidase 10 A_23_P48570 2.36x10-5 5.48 DHRS2 dehydrogenase A_23_P125278 3.03x10-3 5.40 CXCL11 C-X-C motif chemokine ligand 11 A_23_P321501 1.63x10-5 5.38 DHRS2 dehydrogenase A_23_P431388 2.27x10-6 5.33 SPOCD1 SPOC domain containing 1 A_24_P20607 5.13x10-4 5.32 CXCL11 C-X-C motif chemokine ligand 11 A_24_P11061 3.70x10-3 5.30 CSAG1 chondrosarcoma associated gene 1 A_23_P87700 1.03x10-4 5.25 MFAP5 microfibrillar associated protein 5 A_23_P150979 1.81x10-2 5.25 MUCL1 mucin like 1 A_23_P1691 2.71x10-8 5.12 MMP1 matrix metallopeptidase 1 A_23_P350005 2.53x10-4 5.12 TRIML2 tripartite motif family like 2 A_24_P303091 1.23x10-3 4.99 CXCL10 C-X-C motif chemokine ligand 10 A_24_P923612 1.60x10-5 4.95 PTHLH parathyroid hormone like hormone A_23_P7313 6.03x10-5 4.94 SPP1 secreted phosphoprotein 1 A_23_P122924 2.45x10-8 4.93 INHBA inhibin A subunit A_32_P155460 6.56x10-3 4.91 PICSAR P38 inhibited cutaneous squamous cell carcinoma associated lincRNA A_24_P686965 8.75x10-7 4.82 SH2D5 SH2 domain containing 5 A_23_P105475 7.74x10-3 4.70 SLCO1B3 solute carrier organic anion transporter family member 1B3 A_24_P85099 4.82x10-5 4.67 HMGA2 high mobility group AT-hook 2 A_24_P101651 -

Keep Your Fingers Off My DNA: Protein-Protein Interactions
1 2 Keep your fingers off my DNA: 3 protein-protein interactions mediated by C2H2 zinc finger domains 4 5 6 a scholarly review 7 8 9 10 11 Kathryn J. Brayer1 and David J. Segal2* 12 13 14 15 16 17 1Department of Pharmacology and Toxicology, College of Pharmacy, University of Arizona, 18 Tucson, AZ, 85721. 19 2Genome Center and Department of Pharmacology, University of California, Davis, CA, 95616. 20 21 22 23 24 *To whom correspondence should be addressed: 25 David J. Segal, Ph.D. 26 University of California, Davis 27 Genome Center/Pharmacology 28 4513 GBSF 29 451 E. Health Sciences Dr. 30 Davis, CA 95616 31 Tel: 530-754-9134 32 Fax: 530-754-9658 33 Email: [email protected] 34 35 36 Running header: C2H2 ZF interactions with proteins 37 38 Keywords: transcription factors, protein-DNA interactions, protein chemistry, structural biology, 39 functional annotations 40 41 Abstract: 154 words 42 Body Text: 5863 words 43 Figures: 9 44 Tables: 5 45 References: 165 46 C2H2 ZF interactions with proteins Brayer and Segal - review 46 ABSTRACT 47 Cys2-His2 (C2H2) zinc finger domains were originally identified as DNA binding 48 domains, and uncharacterized domains are typically assumed to function in DNA binding. 49 However, a growing body of evidence suggests an important and widespread role for these 50 domains in protein binding. There are even examples of zinc fingers that support both DNA and 51 protein interactions, which can be found in well-known DNA-binding proteins such as Sp1, 52 Zif268, and YY1. C2H2 protein-protein interactions are proving to be more abundant than 53 previously appreciated, more plastic than their DNA-binding counterparts, and more variable and 54 complex in their interactions surfaces. -

Genome-Wide Profiling of Druggable Active Tumor Defense Mechanisms to Enhance Cancer Immunotherapy
bioRxiv preprint doi: https://doi.org/10.1101/843185; this version posted November 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Genome-wide profiling of druggable active tumor defense mechanisms to enhance cancer immunotherapy Rigel J. Kishton1,2,*,#, Shashank J. Patel1,2,†,*, Suman K. Vodnala1,2, Amy E. Decker3, Yogin Patel1,2, Madhusudhanan Sukumar1,2, Tori N. Yamamoto1,2,4, Zhiya Yu1,2, Michelle Ji1,2, Amanda N. Henning1,2, Devikala Gurusamy1,2, Douglas C. Palmer1,2, Winifred Lo1, Anna Pasetto1, Parisa Malekzadeh1, Drew C. Deniger1, Kris C. Wood3, Neville E. Sanjana5,6, Nicholas P. Restifo1,2, #, § 1Surgery Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD 20892, USA 2Center for Cell-Based Therapy, National Cancer Institute, Bethesda, MD 20892, USA 3Department of Pharmacology & Cancer Biology, Duke University School of Medicine, Durham, NC, USA 4Immunology Graduate Group, University of Pennsylvania, Philadelphia, PA 19104, USA 5New York Genome Center, New York, NY 10013 USA 6Department of Biology, New York University, New York, NY 10003, USA *These authors contributed equally to this work. †Present address: NextCure Inc., Beltsville, MD 20705, USA §Present address: Lyell Immunopharma, South San Francisco, CA 94080, USA #Corresponding authors. NPR: [email protected]. RJK: [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/843185; this version posted November 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. -

Genes ZNF350/ZBRK1 and BRIP1/BACH1 Among
HUMAN MUTATION 22:121^128 (2003) RESEARCH ARTICLE Mutational Analysis of the BRCA1-Interacting Genes ZNF350/ZBRK1 and BRIP1/BACH1 Among BRCA1 and BRCA2-Negative Probands From Breast-Ovarian Cancer Families and Among Early-Onset Breast Cancer Cases and Reference Individuals Joni L. Rutter,1 Amelia M. Smith,1 Michael R. Da´vila,1 Alice J. Sigurdson,2 Ruthann M. Giusti,3 Marbin A. Pineda,1 Michele M. Doody,2 Margaret A. Tucker,4 Mark H. Greene,3 Jinghui Zhang,1 and Jeffery P. Struewing1n 1Laboratory of Population Genetics, Center for Cancer Research, National Cancer Institute, Bethesda, Maryland; 2Radiation Epidemiology Branch, Division of Cancer Epidemiology and Genetics; National Cancer Institute, Bethesda, Maryland; 3Clinical Genetics Branch, Division of Cancer Epidemiology and Genetics; National Cancer Institute, Bethesda, Maryland; 4Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics; National Cancer Institute, Bethesda, Maryland Communicated by Mark H. Paalman Two potential breast cancer susceptibility genes, encoding the BRCA1-interacting proteins ZNF350 (or ZBRK1) and BRIP1 (or BACH1), have been identified in yeast two-hybrid screens. We sequenced these genes in probands from 21 families with potentially inherited breast/ovarian cancer, all of which were negative for BRCA1/BRCA2 mutations. Families had at least one case of male breast cancer, two cases of ovarian cancer, or three or more cases of breast and ovarian cancer. In addition, 58 early-onset (before age 35) breast cancer cases and 30 reference individuals were analyzed. Of 17 variants detected in ZBRK1, a missense mutation Val524Ile was identified in the proband of one high-risk family, but no other family members were available for testing. -

Program in Human Neutrophils Fails To
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 is online at: average * The Journal of Immunology Anaplasma phagocytophilum , 20 of which you can access for free at: 2005; 174:6364-6372; ; from submission to initial decision 4 weeks from acceptance to publication J Immunol doi: 10.4049/jimmunol.174.10.6364 http://www.jimmunol.org/content/174/10/6364 Insights into Pathogen Immune Evasion Mechanisms: Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils Dori L. Borjesson, Scott D. Kobayashi, Adeline R. Whitney, Jovanka M. Voyich, Cynthia M. Argue and Frank R. DeLeo cites 28 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2005/05/03/174.10.6364.DC1 This article http://www.jimmunol.org/content/174/10/6364.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* • Why • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Insights into Pathogen Immune Evasion Mechanisms: Anaplasma phagocytophilum Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils1 Dori L. -

Epigenetics of Aging and Alzheimer's Disease
Review Epigenetics of Aging and Alzheimer’s Disease: Implications for Pharmacogenomics and Drug Response Ramón Cacabelos 1,2,* and Clara Torrellas 1,2 Received: 30 September 2015; Accepted: 8 December 2015; Published: 21 December 2015 Academic Editor: Sabrina Angelini 1 EuroEspes Biomedical Research Center, Institute of Medical Science and Genomic Medicine, 15165-Bergondo, Corunna, Spain; [email protected] 2 Chair of Genomic Medicine, Camilo José Cela University, 28692-Madrid, Spain * Correspondence: [email protected]; Tel.: +34-981-780505 Abstract: Epigenetic variability (DNA methylation/demethylation, histone modifications, microRNA regulation) is common in physiological and pathological conditions. Epigenetic alterations are present in different tissues along the aging process and in neurodegenerative disorders, such as Alzheimer’s disease (AD). Epigenetics affect life span and longevity. AD-related genes exhibit epigenetic changes, indicating that epigenetics might exert a pathogenic role in dementia. Epigenetic modifications are reversible and can potentially be targeted by pharmacological intervention. Epigenetic drugs may be useful for the treatment of major problems of health (e.g., cancer, cardiovascular disorders, brain disorders). The efficacy and safety of these and other medications depend upon the efficiency of the pharmacogenetic process in which different clusters of genes (pathogenic, mechanistic, metabolic, transporter, pleiotropic) are involved. Most of these genes are also under the influence of the epigenetic machinery. The information available on the pharmacoepigenomics of most drugs is very limited; however, growing evidence indicates that epigenetic changes are determinant in the pathogenesis of many medical conditions and in drug response and drug resistance. Consequently, pharmacoepigenetic studies should be incorporated in drug development and personalized treatments. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

ZNF263 Is a Transcriptional Regulator of Heparin and Heparan Sulfate Biosynthesis
ZNF263 is a transcriptional regulator of heparin and heparan sulfate biosynthesis Ryan J. Weissa,1, Philipp N. Spahnb,1, Alejandro Gómez Toledoa, Austin W. T. Chiangb, Benjamin P. Kellmanb,JingLia, Christopher Bennerc, Christopher K. Glassa,c,PhilipL.S.M.Gordtsc,d,NathanE.Lewisb,d,e,2, and Jeffrey D. Eskoa,d,2,3 aDepartment of Cellular and Molecular Medicine, University of California San Diego, La Jolla, CA 92093-0687; bDepartment of Pediatrics, University of California San Diego, La Jolla, CA 92093-0760; cDepartment of Medicine, University of California San Diego, La Jolla, CA 92093-0687; dGlycobiology Research and Training Center, University of California San Diego, La Jolla, CA 92093-0687; and eDepartment of Bioengineering, University of California San Diego, La Jolla, CA 92093-0687 Edited by Tadatsugu Taniguchi, University of Tokyo, Meguro-ku, Japan, and approved March 9, 2020 (received for review December 2, 2019) Heparin is the most widely prescribed biopharmaceutical in pro- inactivate thrombin and Factor Xa, which accounts for its potent duction globally. Its potent anticoagulant activity and safety makes anticoagulant activity (4). it the drug of choice for preventing deep vein thrombosis and In 2008, the US Food and Drug Administration issued a major pulmonary embolism. In 2008, adulterated material was intro- recall of pharmaceutical heparin due to contamination of the duced into the heparin supply chain, resulting in several hundred raw heparin stock imported from China. This crisis prompted deaths and demonstrating the need for alternate sources of heparin. new guidelines for monitoring the purity of heparin, but the Heparin is a fractionated form of heparan sulfate derived from feedstock remains vulnerable to natural variation, susceptibility animal sources, predominantly from connective tissue mast cells in of the pig population to infectious agents, and potential con- pig mucosa. -
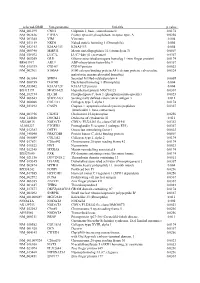
Supplementary Figures S1-S3
selected-GBID Uni-genename Uni-title p value NM_001299 CNN1 Calponin 1, basic, smooth muscle 0.0174 NM_002836 PTPRA Protein tyrosine phosphatase, receptor type, A 0.0256 NM_003380 VIM Vimentin 0.004 NM_033119 NKD1 Naked cuticle homolog 1 (Drosophila) 0.004 NM_052913 KIAA1913 KIAA1913 0.004 NM_005940 MMP11 Matrix metallopeptidase 11 (stromelysin 3) 0.0069 NM_018032 LUC7L LUC7-like (S. cerevisiae) 0.0367 NM_005269 GLI1 Glioma-associated oncogene homolog 1 (zinc finger protein) 0.0174 BE463997 ARL9 ADP-ribosylation factor-like 9 0.0367 NM_015939 CGI-09 CGI-09 protein 0.0023 NM_002961 S100A4 S100 calcium binding protein A4 (calcium protein, calvasculin, 0.0324 metastasin, murine placental homolog) NM_003014 SFRP4 Secreted frizzled-related protein 4 0.0005 NM_080759 DACH1 Dachshund homolog 1 (Drosophila) 0.004 NM_053042 KIAA1729 KIAA1729 protein 0.004 BX415194 MGC16121 Hypothetical protein MGC16121 0.0367 NM_182734 PLCB1 Phospholipase C, beta 1 (phosphoinositide-specific) 0.0023 NM_006643 SDCCAG3 Serologically defined colon cancer antigen 3 0.011 NM_000088 COL1A1 Collagen, type I, alpha 1 0.0174 NM_033292 CASP1 Caspase 1, apoptosis-related cysteine peptidase 0.0367 (interleukin 1, beta, convertase) NM_003956 CH25H Cholesterol 25-hydroxylase 0.0256 NM_144658 DOCK11 Dedicator of cytokinesis 11 0.011 AK024935 NODATA CDNA: FLJ21283 fis, clone COL01910 0.0363 AL050227 PTGER3 Prostaglandin E receptor 3 (subtype EP3) 0.0367 NM_012383 OSTF1 Osteoclast stimulating factor 1 0.0023 NM_145040 PRKCDBP Protein kinase C, delta binding protein 0.0069 NM_000089