High Accuracy Measurement of Isotope Ratios of Molybdenum in Some Terrestrial Molybdenites
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Isotope Production Potential at Sandia National Laboratories: Product, Waste, Packaging, and Transportation*
Isotope Production Potential at Sandia National Laboratories: Product, Waste, Packaging, and Transportation* A. J. Trennel Transportation Systems Department *- *-, o / /"-~~> Sandia National Laboratories** ' J Albuquerque, NM 87185 O Q T » Abstract The U.S. Congress directed the U.S. Department of Energy to establish a domestic source of molybdenum-99, an essential isotope used in nuclear medicine and radiopharmacology. An Environmental Impact Statement for production of 99Mo at one of four candidate sites is being prepared. As one of the candidate sites, Sandia National Laboratories is developing the Isotope Production Project. Using federally approved processes and procedures now owned by the U.S. Department of Energy, and existing facilities that would be modified to meet the production requirements, the Sandia National Laboratories' Isotope Project would manufacture up to 30 percent of the U.S. market, with the capacity to meet 100 percent of the domestic need if necessary. This paper provides a brief overview of the facility, equipment, and processes required to produce isotopes. Packaging and transportation issues affecting both product and waste are addressed, and the storage and disposal of the four low-level radioactive waste types generated by the production program are considered. Recommendations for future development are provided. This work was performed at Sandia National Laboratories, Albuquerque, New Mexico, for the U.S. Department of Energy under Contract DE-AC04-94AL85000. A U.S. Department of Energy facility. DISTRPJTO OF THIS DOCUMENT IS UNLIMITED #t/f W A8 1 fcll PROJECT NEED AND BACKGROUND Nuclear medicine is an expanding segment of today's medical and pharmaceutical communities. Specific radioactive isotopes are vital, with molybdenum-99 (99Mo) being the most important medical isotope. -
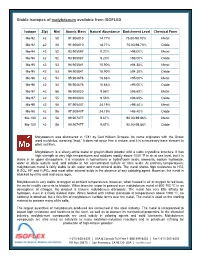
Stable Isotopes of Molybdenum Available from ISOFLEX
Stable isotopes of molybdenum available from ISOFLEX Isotope Z(p) N(n) Atomic Mass Natural Abundance Enrichment Level Chemical Form Mo-92 42 50 91.906810 14.77% 75.00-98.70% Metal Mo-92 42 50 91.906810 14.77% 75.00-98.70% Oxide Mo-94 42 52 93.905087 9.23% >98.00% Metal Mo-94 42 52 93.905087 9.23% >98.00% Oxide Mo-95 42 53 94.905841 15.90% ≥94.30% Metal Mo-95 42 53 94.905841 15.90% ≥94.30% Oxide Mo-96 42 54 95.904678 16.68% >95.00% Metal Mo-96 42 54 95.904678 16.68% >95.00% Oxide Mo-97 42 55 96.906020 9.56% ≥96.60% Metal Mo-97 42 55 96.906020 9.56% ≥96.60% Oxide Mo-98 42 56 97.905407 24.19% >98.40% Metal Mo-98 42 56 97.905407 24.19% >98.40% Oxide Mo-100 42 58 99.907477 9.67% 90.00-99.86% Metal Mo-100 42 58 99.907477 9.67% 90.00-99.86% Oxide Molybdenum was discovered in 1781 by Carl William Scheele. Its name originates with the Greek word molybdos, meaning “lead.” It does not occur free in nature, and it is a necessary trace element in plant nutrition. Molybdenum is a silvery-white metal or grayish-black powder with a cubic crystalline structure. It has high strength at very high temperatures and oxidizes rapidly above 1000 °F in air at sea level, but it is stable in an upper atmosphere. -

PSI • Scientific Report 1999 /Volume I
CH0000002 PAUL SCHERRER INSTITUT ISSN 1423-7296 March 2000 PSI • Scientific Report 1999 /Volume I Particles and Matter 3 1/28 An international collaboration of radiochemists led by the Laboratory for Radiochemistry and Environmental Chemistry of PSl and Bern University succeeded for the first time to experimentally investigate the chemical properties of bohrium (element 107) and to establish it as a member of group 7 of the Periodic Table. During a one-month long experiment, a total of only six bohrium atoms were gaschromatographically isolated in the form of volatile bohrium oxychloride molecules and identified by registering their unique decay sequence of alpha particle emissions via dubnium (element 105) to lawrencium (element 103). The short-lived bohrium atoms, decaying with a half-life of about 20 s, were produced by bombarding a very rare, highly radioactive berkelium target supplied by the US department of energy with an intense beam of neon ions at the PSl injector 1 cyclotron. PAUL SCHERRER INSTITUT ISSN 1423-7296 March 2000 \} Scientific Report 1999 Volume I Particles and Matter ed. by: J. Gobrecht, H. Gaggeler, D. Herlach, K. Junker, P.-R. Kettle, P. Kubik, A. Zehnder CH-5232 Villigen PSI Switzerland Telephone:+41 56 310 21 11 Telefax:+ 41 56 310 21 99 http://www.psi.ch TABLE OF CONTENTS Introduction .1 Laboratory for Particle Physics 3 Foreword 4 Particle Physics Theory Theory (I) 5 Theory (II) 6 Ring Accelerator Experiments Particle Properties and Decays A precise measurement of the Jt+-^7t°e+v decay rate 7 Measurement of -

Molybdenum in the Open Cluster Stars
MOLYBDENUM IN THE OPEN CLUSTER STARS T. Mishenina1, E. Shereta1, M. Pignatari2,3,6,7, G. Carraro4, T. Gorbaneva1, C. Soubiran5 1Astronomical Observatory, Odessa National University, Marazlievskaia Str., 1V, 65014, Odessa, Ukraine [email protected] 2 E.A. Milne Centre for Astrophysics, Department of Physics & Mathematics, University of Hull, HU6 7RX, United Kingdom [email protected] 3 Konkoly Observatory, Hungarian Academy of Sciences, Konkoly Thege Miklos ut 15-17, H-1121 Budapest, Hungary 4 Dipartimento di Fisica e Astronomia, Universita di Padova, I-35122, Padova, Italy [email protected] 5 Laboratoire d’astrophysique de Bordeaux, Universit´eBordeaux, CNRS, B18N, all´ee Geoffroy Saint-Hilaire, 33615 Pessac, France [email protected] 6 Joint Institute for Nuclear Astrophysics - Center for the Evolution of the Elements, USA 7 NuGrid Collaboration https://nugrid.github.io/ June 9, 2020 arXiv:2006.04629v1 [astro-ph.GA] 8 Jun 2020 Abstract Molybdenum abundances in the stars from 13 different open clusters were determined. High-resolution stellar spectra were obtained using the VLT tele- scope equipped with the UVES spectrograph on Cerro Paranal, Chile. The Mo abundances were derived in the LTE approximation from the Mo I lines at 5506 A˚ and 5533 A.˚ A comparative analysis of the behaviour of molyb- denum in the sampled stars of open clusters and Galactic disc show similar trends of decreasing Mo abundances with increasing metallicities; such a be- haviour pattern suggests a common origin of the examined populations. On the other hand, the scatter of Mo abundances in the open cluster stars is slightly greater, 0.14 dex versus 0.11 dex. -

Calcium Isotopes in Natural and Experimental Carbonated Silicate Melts
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 2-27-2018 2:30 PM Calcium Isotopes in Natural and Experimental Carbonated Silicate Melts Matthew Maloney The University of Western Ontario Supervisor Bouvier, Audrey The University of Western Ontario Co-Supervisor Withers, Tony The University of Western Ontario Graduate Program in Geology A thesis submitted in partial fulfillment of the equirr ements for the degree in Master of Science © Matthew Maloney 2018 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Geochemistry Commons Recommended Citation Maloney, Matthew, "Calcium Isotopes in Natural and Experimental Carbonated Silicate Melts" (2018). Electronic Thesis and Dissertation Repository. 5256. https://ir.lib.uwo.ca/etd/5256 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. Abstract The calcium stable isotopic compositions of mantle-sourced rocks and minerals were investigated to better understand the carbon cycle in the Earth’s mantle. Bulk carbonatites and kimberlites were analyzed to identify a geochemical signature of carbonatite magmatism, while inter-mineral fractionation was measured in co-existing Ca-bearing carbonate and silicate minerals. Bulk samples show a range of composition deviating from the bulk silicate Earth δ44/40Ca composition indicating signatures of magmatic processes or marine carbonate addition 44/40 to source materials. Δ Cacarbonate-silicate values range from -0.55‰ to +1.82‰ and positively correlate with Ca/Mg ratios in pyroxenes. -

PRODUCTION of Mo-99/Tc-99M VIA PHOTONEUTRON REACTION USING
PRODUCTION OF Mo-99/Tc-99m VIA PHOTONEUTRON REACTION USING NATURAL MOLYBDENUM A Thesis by TALAL BADER HARAHSHEH Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Chair of Committee, Gamal Akabani Co-Chair of Committee, John W. Poston, Sr. Committee Member, Victor M. Ugaz Head of Department, Yassin A. Hassan May 2017 Major Subject: Nuclear Engineering Copyright 2017 Talal Harahsheh ABSTRACT The current shortage of the widely used radionuclide 99mTc has raised many concerns in the nuclear medicine industry. The shortage is caused by the outdated method of production using highly enriched uranium (HEU) and nuclear reactors. This production method is no longer feasible due to the restrictions on the use of HEU. Alternative methods of production must be addressed to ensure the uninterrupted supply of this key radionuclide. This research analyzes an alternative production method of 99mTc using an accelerator-generated 99Mo, which is the parent element of 99mTc. The radionuclide 99mTc has a short half-life of 6 hours. However, 99Mo, the parent nuclide has a much longer half-life of 66 hours. Usually, a medical facility receives a 99Mo/99mTc generator. Technetium-99m would be obtained from the 99Mo provided through chemical separation, which is conducted at the medical facilities. The main issues to be addressed include the production of 99Mo using the photonuclear reaction, the containment and exclusion of unwanted decay products, ensuring the integrity of the end-product 99Mo/99mTc post chemical separation, and assessing the economic feasibility of the complete strategy, including life cycle. -

Use of Stable Isotopes and Mathematical Modelling to Investigate Human Mineral Metabolism
Nutrition Research Reviews (2001), 14, 295–315 DOI: 10.1079/NRR200124 © The Author 2001 Use of stable isotopes and mathematical modelling to investigate human mineral metabolism Jack R. Dainty Institute of Food Research, Norwich NR4 7UA, UK Individuals have varying needs for minerals that are dependent, amongst other things, on their lifestyle, age and genetic makeup. Knowledge of exact individual nutritional requirements should lead to better health, increased quality of life and reduced need for expensive medical care. Bioavailability, nutrient–gene interactions and whole-body metabolism all need to be investigated further if we are to progress towards the goal of defining optimal health and nutritional status. The discussion which fol- lows will critically review the latest developments in the area of metabo- lism for several of the minerals that are essential for human health: Ca, Zn, Cu and Se. Stable-isotope tracers and mathematical modelling are some of the tools being used to facilitate the greater understanding in uptake, utilisation and excretion of these minerals. Stable isotopes, admin- istered in physiological doses, present little or no risk to volunteers and allow metabolic studies to be carried out in vulnerable population groups such as children and pregnant women. Intrinsic labelling of foodstuffs ensures that the tracer and the native mineral will behave similarly once inside the body. Advances in computing power and software dedicated to solving nutritional problems have made it possible for investigators to use mathematical modelling in their experimental work. Mineral metabolism is ideally suited to a form of modelling known as compartmental analysis, which allows rates of mineral transfer and sizes of mineral stores to be calculated accurately without the need for invasive sampling of body tissues. -

Australian Atomic Energy Commission Research Establishment
AAEC/E644 AUSTRALIAN ATOMIC ENERGY COMMISSION RESEARCH ESTABLISHMENT LUCAS HEIGHTS RESEARCH LABORATORIES EVALUATION OF NEUTRON ACTIVATION ANALYSIS FOR THE MEASUREMENT OF ISOTOPIC ABUNDANCES IN MOLYBDENUM-98 ENRICHED MOLYBDENUM by E. L. R. HETHERINGTON MARCH 1987 ISBN 0 642 59847 9 AUSTRALIAN ATOMIC ENERGY COMMISSION RESEARCH ESTABLISHMENT LUCAS HEIGHTS RESEARCH LABORATORIES EVALUATION OF NEUTRON ACTIVATION ANALYSIS FOR THE MEASUREMENT OF ISOTOPIC ABUNDANCES IN MOLYBDENUM-98 ENRICHED MOLYBDENUM by E.L.R. HETHERINGTON ABSTRACT Molybdenum enriched to approximately 50 per cent 98Mo is required for the development of a portable 99mTc generator based on the zirconium molybdate gel system developed by the AAEC. It has been proposed that existing AAEC facilities be used to enrich molybdenum, and neutron activation analysis has been evaluated as a means of monitoring the enrichment process. 98 Simulated enriched targets of Mo03 with Mo abundances ranging from 30 to 90 per cent were prepared by 98 diluting 96.87 per cent enriched MoO3 with the natural trioxide. These targets were each irradiated 12 2 1 simultaneously with a reference natural Mo03 target in a thermal neutron flux of 7.5 x 10 neutrons cm~ s~ where the reactions 92Mo(n/y) 93Mo, 98Mo(n,Y) 99Mo and 100Mo(n,Y)101Mo-101Tc occur. The isotopic abundances of 98Mo and 100Mo were determined with an accuracy ± 2 per cent by comparing the yields of 99Mo-99mTc and 101Mo-101Tc, respectively, in the enriched and natural targets. The activation yield of 93Mo was too low for the determination of the 92Mo abundances. National Library of Australia card number and ISBN 0 642 59847 9 The following descriptors have been selected from the INIS Thesaurus to describe the subject content of this report for Information retrieval purposes. -

A Link Between Oxygen, Calcium and Titanium Isotopes in 26Al-Poor
Available online at www.sciencedirect.com ScienceDirect Geochimica et Cosmochimica Acta 189 (2016) 70–95 www.elsevier.com/locate/gca A link between oxygen, calcium and titanium isotopes in 26Al-poor hibonite-rich CAIs from Murchison and implications for the heterogeneity of dust reservoirs in the solar nebula Levke Ko¨o¨p a,b,c,⇑, Andrew M. Davis a,b,c,d, Daisuke Nakashima e,f, Changkun Park g,h, Alexander N. Krot g, Kazuhide Nagashima g, Travis J. Tenner e, Philipp R. Heck a,b,c, Noriko T. Kita e a Department of the Geophysical Sciences, The University of Chicago, Chicago, IL, USA b Chicago Center for Cosmochemistry, The University of Chicago, Chicago, IL, USA c Robert A. Pritzker Center for Meteoritics and Polar Studies, Field Museum of Natural History, Chicago, IL, USA d Enrico Fermi Institute, The University of Chicago, Chicago, IL, USA e Dept. Geoscience, University of Wisconsin-Madison, WI, USA f Division of Earth and Planetary Materials Science, Tohoku University, Sendai, Japan g HIGP/SOEST University of Hawai‘i at Manoa, Honolulu, HI, USA h Korea Polar Research Institute, Incheon, Republic of Korea Received 30 July 2015; accepted in revised form 6 May 2016; available online 13 May 2016 Abstract PLACs (platy hibonite crystals) and related hibonite-rich calcium-, aluminum-rich inclusions (CAIs; hereafter collectively referred to as PLAC-like CAIs) have the largest nucleosynthetic isotope anomalies of all materials believed to have formed in the solar system. Most PLAC-like CAIs have low inferred initial 26Al/27Al ratios and could have formed prior to injection or widespread distribution of 26Al in the solar nebula. -
![Arxiv:1901.06959V1 [Physics.Atom-Ph] 21 Jan 2019 Some Care, Understood As a Potential Signature of NP](https://docslib.b-cdn.net/cover/0721/arxiv-1901-06959v1-physics-atom-ph-21-jan-2019-some-care-understood-as-a-potential-signature-of-np-2120721.webp)
Arxiv:1901.06959V1 [Physics.Atom-Ph] 21 Jan 2019 Some Care, Understood As a Potential Signature of NP
The g factor of bound electrons as a test for physics beyond the Standard Model V. Debierre,∗ C. H. Keitel, and Z. Harmany Max Planck Institute for Nuclear Physics, Saupfercheckweg 1, 69117 Heidelberg The use of high-precision measurements of the g factor of few-electron ions and its isotope shifts is put forward as a probe for physics beyond the Standard Model. The contribution of a hypothetical fifth fundamental force to the g factor is calculated for the ground state of H-like, Li-like and B-like ions, and employed to derive bounds on the parameters of that force. The weighted difference and especially the isotope shift of g factors are used in order to increase the experimental sensitivity to the new physics contribution. It is found that, combining measurements from four different isotopes of H-like, Li-like and B-like calcium ions at currently accessible accuracy levels, experimental results compatible with King planarity would constrain the new physics coupling constant more than one order of magnitude further than the best current atomic data. The g factors of the free electon and free muon have boson.|New scalar bosons have been proposed as a so- served as precision tests for quantum electrodynamics lution to the long-standing electroweak hierarchy prob- (QED), the Standard Model (SM) more broadly, and pos- lem [36]. These massive scalar bosons would carry a fifth sible extensions of the SM [1{7]. In parallel, recent years force, resulting in an interaction between neutrons and have seen rapid improvements in the experimental [8{12] electrons, by coupling to both particle types in a spin- and theoretical [13{21] determination of the g factor of independent way [32, 33, 35]. -

Potential Production of Molybdenum-99
DEVELOPMENT OF A 37-ELEMENT FUEL BUNDLE FOR THE PRODUCTION OF MOLYBDENUM-99 IN CANDU POWER REACTORS By Jawad Haroon A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Applied Science In The Faculty of Energy Systems and Nuclear Science Nuclear Engineering University of Ontario Institute of Technology August 2014 © Jawad Haroon, 2014 Abstract In this study, the potential use of CANDU power reactors for the production of Mo-99 is assessed. Five different modifications of a 37-element fuel bundle that could be used for the production of Mo-99 in existing CANDU type power reactors are explored. Since Mo-99 is generated by the fission of U-235 with a fission yield of 6.1%, the proposed designs use enriched uranium in the Mo-99 producing fuel pins. The challenge lies in designing a fuel that is able to produce significant quantities of Mo-99 while having similar neutronics and thermalhydraulic characteristics to the standard CANDU fuel. The proposed designs, when irradiated in the peak power channel of a CANDU core, are shown to produce significant quantities of Mo-99 while maintaining the necessary reactivity and power rating limits. The total Mo-99 production activities are found to be approximately 2335, 2297, 2336, 4131 and 4268 six-day Curies per bundle for Designs 1 to 5, respectively. The yield corresponds to approximately 19% (for Designs 1, 2 and 3) and 34% (for Designs 4 and 5) of the world weekly demand for Mo-99. A production cycle of 6 bundles per week (for Designs 1, 2 and 3), or 3 bundles per week (for Designs 4 and 5) can meet the global demand of Mo-99 medical isotopes. -

Medical Uses of Radioactive Calcium Review of an Iaea Programme to Promote the Applications of Calcium-47
between 0.1 andO. 15 mill/kWh of nuclear electricity disposal operation for calcined solids in salt deposits produced. This cost approximates only one to two are estimated at 0-01 mill/kWh(e). Based on past per cent of the cost of nuclear power in an 8 - 10 laboratory and engineering scale cold unit operation mill/kWh economy. The ORNL engineering and data, and on an expected successful field demonstra economic studies indicate that the total cost of in tion and testing programme with high activity wastes, terim liquid storage, pot calcination, and shipping it is firmly believed that waste management opera over a 3000 mile round trip can be as low as tions should not constitute a major obstacle to the 0-02 mill/kWh(e). Tentative costs for a large-scale development of economical nuclear power. " MEDICAL USES OF RADIOACTIVE CALCIUM REVIEW OF AN IAEA PROGRAMME TO PROMOTE THE APPLICATIONS OF CALCIUM-47 Calcium plays a number of biologically essen absorbed in a layer of matter less than 1 mm thick. tial roles, which have long been under investigation The relatively long half-life of the isotope results, in by various techniques available to medical science. most human studies, in an undesirably prolonged One of the most important of these techniques is irradiation of the subject and the isotope is there radioactive tracer analysis, i. e. study of the func fore considered rather hazardous for internal ad tions of calcium within the body with the help of a ministration. Also, the low energy level of its radia radioactive isotope of the element.