Overpotential and Catalytic Activity. by Erick
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Towards the Hydrogen Economy—A Review of the Parameters That Influence the Efficiency of Alkaline Water Electrolyzers
energies Review Towards the Hydrogen Economy—A Review of the Parameters That Influence the Efficiency of Alkaline Water Electrolyzers Ana L. Santos 1,2, Maria-João Cebola 3,4,5 and Diogo M. F. Santos 2,* 1 TecnoVeritas—Serviços de Engenharia e Sistemas Tecnológicos, Lda, 2640-486 Mafra, Portugal; [email protected] 2 Center of Physics and Engineering of Advanced Materials (CeFEMA), Instituto Superior Técnico, Universidade de Lisboa, 1049-001 Lisbon, Portugal 3 CBIOS—Center for Research in Biosciences & Health Technologies, Universidade Lusófona de Humanidades e Tecnologias, Campo Grande 376, 1749-024 Lisbon, Portugal; [email protected] 4 CERENA—Centre for Natural Resources and the Environment, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisbon, Portugal 5 Escola Superior Náutica Infante D. Henrique, 2770-058 Paço de Arcos, Portugal * Correspondence: [email protected] Abstract: Environmental issues make the quest for better and cleaner energy sources a priority. Worldwide, researchers and companies are continuously working on this matter, taking one of two approaches: either finding new energy sources or improving the efficiency of existing ones. Hydrogen is a well-known energy carrier due to its high energy content, but a somewhat elusive one for being a gas with low molecular weight. This review examines the current electrolysis processes for obtaining hydrogen, with an emphasis on alkaline water electrolysis. This process is far from being new, but research shows that there is still plenty of room for improvement. The efficiency of an electrolyzer mainly relates to the overpotential and resistances in the cell. This work shows that the path to better Citation: Santos, A.L.; Cebola, M.-J.; electrolyzer efficiency is through the optimization of the cell components and operating conditions. -

Electrode Reactions and Kinetics
CHEM465/865, 2004-3, Lecture 14-16, 20 th Oct., 2004 Electrode Reactions and Kinetics From equilibrium to non-equilibrium : We have a cell, two individual electrode compartments, given composition, molar reaction Gibbs free ∆∆∆ energy as the driving force, rG , determining EMF Ecell (open circuit potential) between anode and cathode! In other words: everything is ready to let the current flow! – Make the external connection through wires. Current is NOT determined by equilibrium thermodynamics (i.e. by the composition of the reactant mixture, i.e. the reaction quotient Q), but it is controlled by resistances, activation barriers, etc Equilibrium is a limiting case – any kinetic model must give the correct equilibrium expressions). All the involved phenomena are generically termed ELECTROCHEMICAL KINETICS ! This involves: Bulk diffusion Ion migration (Ohmic resistance) Adsorption Charge transfer Desorption Let’s focus on charge transfer ! Also called: Faradaic process . The only reaction directly affected by potential! An electrode reaction differs from ordinary chemical reactions in that at least one partial reaction must be a charge transfer reaction – against potential-controlled activation energy, from one phase to another, across the electrical double layer. The reaction rate depends on the distributions of species (concentrations and pressures), temperature and electrode potential E. Assumption used in the following: the electrode material itself (metal) is inert, i.e. a catalyst. It is not undergoing any chemical transformation. It is only a container of electrons. The general question on which we will focus in the following is: How does reaction rate depend on potential? The electrode potential E of an electrode through which a current flows differs from the equilibrium potential Eeq established when no current flows. -

Lecture 03 Electrochemical Kinetics
EMA4303/5305 Electrochemical Engineering Lecture 03 Electrochemical Kinetics Dr. Junheng Xing, Prof. Zhe Cheng Mechanical & Materials Engineering Florida International University Electrochemical Kinetics Electrochemical kinetics “Electrochemical kinetics is a field of electrochemistry studying the rate of electrochemical processes. Due to electrochemical phenomena unfolding at the interface between an electrode and an electrolyte, there are accompanying phenomena to electrochemical reactions which contribute to the overall reaction rate.” (Wikipedia) “The main goal of the electrochemical kinetics is to find a relationship between the electrode overpotential and current density due to an applied potential.” (S. N. Lvov) Main contents of this lecture . Overpotential (η) . Charge transfer overpotential − Butler-Volmer equation − Tafel equation . Mass transfer overpotential EMA 5305 Electrochemical Engineering Zhe Cheng (2017) 3 Electrochemical Kinetics 2 Cell Potential in Different Modes Electrolytic cell (EC) Galvanic cell (GC) 푬푬푸 = 푰 (푹풆풙풕 + 푹풊풏풕) 푬푨풑풑풍풊풆풅 = 푬푬푸 + 푰푹풊풏풕 푬풆풙풕 = 푬푬푸 − 푰푹풊풏풕= IRext EMA 5305 Electrochemical Engineering Zhe Cheng (2017) 3 Electrochemical Kinetics 3 Cell Potential-Current Dependence . 푹풊풏풕 represents the total internal resistance of the cell, which is usually not zero, in most cases. Therefore, the difference between cell equilibrium potential and apparent potential is proportional to the current passing through the cell. Cell potential-current dependences EMA 5305 Electrochemical Engineering Zhe Cheng (2017) 3 Electrochemical Kinetics 4 Overpotential (η) Overpotential for a single electrode . The difference between the actual potential (measured) for an electrode, E, and the equilibrium potential for that electrode, 푬푬푸, is called the overpotential of that electrode, 휼 = 푬 −푬푬푸, which could be either positive or negative, depending on the direction of the half (cell) reaction. -

Solar-Driven, Highly Sustained Splitting of Seawater Into Hydrogen and Oxygen Fuels
Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels Yun Kuanga,b,c,1, Michael J. Kenneya,1, Yongtao Menga,d,1, Wei-Hsuan Hunga,e, Yijin Liuf, Jianan Erick Huanga, Rohit Prasannag, Pengsong Lib,c, Yaping Lib,c, Lei Wangh,i, Meng-Chang Lind, Michael D. McGeheeg,j, Xiaoming Sunb,c,d,2, and Hongjie Daia,2 aDepartment of Chemistry, Stanford University, Stanford, CA 94305; bState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China; cBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China; dCollege of Electrical Engineering and Automation, Shandong University of Science and Technology, Qingdao 266590, China; eDepartment of Materials Science and Engineering, Feng Chia University, Taichung 40724, Taiwan; fStanford Synchrotron Radiation Light Source, SLAC National Accelerator Laboratory, Menlo Park, CA 94025; gDepartment of Materials Science and Engineering, Stanford University, Stanford, CA 94305; hCenter for Electron Microscopy, Institute for New Energy Materials, Tianjin University of Technology, Tianjin 300384, China; iTianjin Key Laboratory of Advanced Functional Porous Materials, School of Materials, Tianjin University of Technology, Tianjin 300384, China; and jDepartment of Chemical Engineering, University of Colorado Boulder, Boulder, CO 80309 Contributed by Hongjie Dai, February 5, 2019 (sent for review January 14, 2019; reviewed by Xinliang Feng and Ali Javey) Electrolysis of water to generate hydrogen fuel is an attractive potential ∼490 mV higher than that of OER, which demands renewable energy storage technology. However, grid-scale fresh- highly active OER electrocatalysts capable of high-current (∼1 2 water electrolysis would put a heavy strain on vital water re- A/cm ) operations for high-rate H2/O2 production at over- sources. -

Overpotential Analysis of Alkaline and Acidic Alcohol Electrolysers And
1 2 Overpotential analysis of alkaline and acidic alcohol electrolysers 3 and optimized membrane-electrode assemblies 4 5 F. M. Sapountzia*, V. Di Palmab, G. Zafeiropoulosc, H. Penchevd, M.Α. Verheijenb, 6 M. Creatoreb, F. Ublekovd, V. Sinigerskyd, W.M. Arnold Bikc, H.O.A. Fredrikssona, 7 M.N. Tsampasc, J.W. Niemantsverdrieta 8 9 a SynCat@DIFFER, Syngaschem BV, P.O. Box 6336, 5600 HH, Eindhoven, The Netherlands, 10 www.syngaschem.com 11 b Department of Applied Physics, Eindhoven University of Technology, 5600MB, Eindhoven, The Netherlands 12 c DIFFER, Dutch Institute For Fundamental Energy Research, De Zaale 20, 5612 AJ, Eindhoven, The Netherlands 13 d Institute of Polymers, Bulgarian Academy of Sciences, Acad. Georgi Bonchev 103A Str., BG 1113, Sofia, 14 Bulgaria 15 16 *Corresponding author: Foteini Sapountzi, email: [email protected], postal address: Syngaschem BV, P.O. 17 Box 6336, 5600 HH, Eindhoven, The Netherlands 18 19 20 Abstract: Alcohol electrolysis using polymeric membrane electrolytes is a promising route for 21 storing excess renewable energy in hydrogen, alternative to the thermodynamically limited water 22 electrolysis. By properly choosing the ionic agent (i.e. H+ or OH-) and the catalyst support, and 23 by tuning the catalyst structure, we developed membrane-electrode-assemblies which are 24 suitable for cost-effective and efficient alcohol electrolysis. Novel porous electrodes were 25 prepared by Atomic Layer Deposition (ALD) of Pt on a TiO2-Ti web of microfibers and were 26 interfaced to polymeric membranes with either H+ or OH- conductivity. Our results suggest that 27 alcohol electrolysis is more efficient using OH- conducting membranes under appropriate 28 operation conditions (high pH in anolyte solution). -

Understanding Anode Overpotential
UNDERSTANDING ANODE OVERPOTENTIAL Rebecca Jayne Thorne1, Camilla Sommerseth1, Ann Mari Svensson1, Espen Sandnes2, Lorentz Petter Lossius2, Hogne Linga2, and Arne Petter Ratvik1 1Dept. of Materials Science and Engineering, Norwegian University of Science and Technology, NO-7491 Trondheim, Norway 2Hydro Aluminium, Årdal, Norway Keywords: Anode, carbon, electrochemistry, characterisation, overpotential. Abstract have not linked these coke properties to overpotential directly [2, 9-11]. It is likely that coke and anode physical properties will In aluminium electrolysis cells the anodic process is associated have an effect on overpotential, as the reactivity of carbons, both with a substantial overpotential. Industrial carbon anodes are electrochemical reactions and oxidation reactions, is generally produced from a blend of coke materials, but the effect of coke known to be related to microstructural parameters [12, 13]. In type on anodic overpotential has not been well studied. In this addition, the formation and release of CO2 bubbles also relies on work, lab-scale anodes were fabricated from single cokes and the surface microstructure [14]. electrochemical methods were subsequently used to determine the overpotential trend of the anode materials. Attempts were then In a previous study that served as preliminary work on a new made to explain these trends in terms of both the physical and project, the total overpotential of a range of carbon anodes chemical characteristics of the baked anodes themselves and their varying only in coke type was measured [15]. These anodes were raw materials. Routine coke and anode characterisation methods labeled 1 to 4, and a distinct overpotential trend was found with were used to measure properties such as impurity concentrations anode 4 having a lower overpotential than anode 1 by -2 and reactivity (to air and CO2), while non-routine characterisation approximately 200 mV at 1 A cm . -
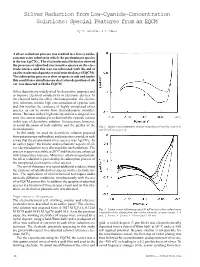
Silver Reduction from Low-Cyanide-Concentration Solutions: Special Features from an EQCM
Silver Reduction from Low-Cyanide-Concentration Solutions: Special Features from an EQCM By H. Sanchez & Y. Meas A silver reduction process was studied in a low-cyanide- concentration solution in which the predominant species - is the ion Ag(CN)2 . The electrochemical behavior showed the presence of adsorbed electroactive species on the elec- trode surface and this was corroborated with the aid of an electrochemical quartz crystal microbalance (EQCM). The adsorption process is slow at open circuit and under this condition a simultaneous electroless deposition of sil- ver was detected with the EQCM. Silver deposits are widely used for decorative purposes and to improve electrical conductivity in electronic devices. In the classical baths for silver electrodeposition, the electro- lytic solutions contain high concentrations of cyanide ions and this implies the existence of highly complexed silver species, as can be shown from thermodynamic consider- ations.1 Because of their high toxicity and for ecological rea- sons, the current tendency is to diminish the cyanide content in this type of electrolytic solution. It is necessary, however, to avoid alteration of bath stability and the quality of the Fig. 1—Single scan voltammetry of silver reduction at 10 mV/sec (curve 1) electrodeposits. and 20 mV/sec (curve 2). In this study, we used an electrolytic solution prepared from potassium pyrophosphate and potassium cyanide in such - a way that the predominant silver species was Ag(CN)2 . In an earlier paper,1 the kinetic and mechanistic aspects of sil- ver electroreduction were discussed for such solutions. The process is quasi-reversible at 20 °C and kinetics are enhanced with temperature increase. -

Role of Diffuse Layer in Acidic and Alkaline Fuel Cells
Author's personal copy Electrochimica Acta 56 (2011) 4518–4525 Contents lists available at ScienceDirect Electrochimica Acta journal homepage: www.elsevier.com/locate/electacta Role of the diffuse layer in acidic and alkaline fuel cells Isaac Sprague, Prashanta Dutta ∗ School of Mechanical and Materials Engineering, Washington State University, Pullman, WA 99164-2920, USA article info abstract Article history: A numerical model is developed to study electrolyte dependent kinetics in fuel cells. The model is based Received 17 December 2010 on the Poisson–Nernst–Planck (PNP) and generalized-Frumkin–Butler–Volmer (gFBV) equations, and is Received in revised form 13 February 2011 used to understand how the diffuse layer and ionic transport play a role in the performance difference Accepted 15 February 2011 between acidic and alkaline systems. The laminar flow fuel cell (LFFC) is used as the model fuel cell Available online 23 February 2011 architecture to allow for the appropriate comparison of equivalent acidic and alkaline systems. We study the overall cell performance and individual electrode polarizations of acidic and alkaline fuel cells for Keywords: both balanced and unbalanced electrode kinetics as well as in the presence of transport limitations. The Alkaline fuel cell Acidic electrolyte results predict cell behavior based on electrolyte composition that strongly correlates with observed Alkaline electrolyte experimental results from literature and provides insight into the fundamental cause of these results. Electric double layer Specifically, it is found that the working ion concentration at the reaction plane plays a significant role in Laminar flow fuel cell fuel cell performance including activation losses and the response to different kinetic rates at an individual electrode. -

Mechanism of the Chlorine Evolution on a Ruthenium Oxide/Titanium Oxide Electrode and on a Ruthenium Electrode
Mechanism of the chlorine evolution on a ruthenium oxide/titanium oxide electrode and on a ruthenium electrode Citation for published version (APA): Janssen, L. J. J., Starmans, L. M. C., Visser, J. G., & Barendrecht, E. (1977). Mechanism of the chlorine evolution on a ruthenium oxide/titanium oxide electrode and on a ruthenium electrode. Electrochimica Acta, 22(10), 1093-1100. https://doi.org/10.1016/0013-4686(77)80045-5 DOI: 10.1016/0013-4686(77)80045-5 Document status and date: Published: 01/01/1977 Document Version: Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers) Please check the document version of this publication: • A submitted manuscript is the version of the article upon submission and before peer-review. There can be important differences between the submitted version and the official published version of record. People interested in the research are advised to contact the author for the final version of the publication, or visit the DOI to the publisher's website. • The final author version and the galley proof are versions of the publication after peer review. • The final published version features the final layout of the paper including the volume, issue and page numbers. Link to publication General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. -

Electroorganic Chemistry: Choice of Electrodes 1
Yu Kawamata Electroorganic Chemistry: Choice of Electrodes Baran Group Meeting 12/10/2016 1 Also see: Electrochemistry in Synthesis (Ambhaikar, 2005) Electroorganic Chemistry (Rosen, 2014) Unique parameters in electrochemistry: Electrochemical cell ••••• devided or undivided Current and potential ••••• constant current (c.c.) or constant potential (c.p.) Electrolyte ••••• various ammonium or alkali metal salts Electrodes ••••• anode (oxidation), cathode (reduction) Divided cell Undivided cell • Anode compartment is always • Net reduction proceeds if a sacrificial Topics not discussed: oxidation. anode is employed. • Chemically modified electrodes • Cathode compartment is always • In other cases, reaction type (net oxidation • Electrodes for inorganic and analytical electrochemistry reduction. or reduction) depends on the balance of redox potentials of compounds in the cell. 1. Electrode material Review: Pletcher, Chem. Rev. 1990, 90, 837. 1-3. Overpotential Overpotential is the potential 1-1. General Consideration difference between a half- i) Physical stability v) cost and lifetime reaction's thermodynamically determined potential and the ii) chemical stability vi) suitable physical form potential at which the redox event iii) overpotential vii) electrical conductivity is experimentally observed. iv) rate and product selecticity Thermodinamically defined potential: 1-2. Modes of action of electrodes Oxygen O + 4H+ + 4e- 2H O º = 1.23 (V) Electrodes can: transfer electrons 2 2 E red (vs SHE) absorb organic compounds Hydrogen + - º 2H + 2e H2 E = 0.00 (V) Reactivity and product selectivity might be red https://en.wikipedia.org/wiki/overpotential affected by electrode materials. act as reagents Generally, material with large O2 overpotential is used for anode and material with large H2 ovarpotential is used for cathode. -

Faradaic Rxns: More General Situations--Occur Reaches Value
Faradaic Rxns: More General Situations--occur when DF WE/Soln reaches value adequate to achieve electron transfer across interface---either reduction or oxidation Faradaic current tells us about the rate of an electro- chemical reaction. Since faradaic current is related to Faraday’s law of electrolysis: N = q/nF = moles electrolyzed at WE rate of this electrochemical reaction-- dN/dt = (dq/dt)/nF = i/nF =Velocity = moles/sec However electrode reactions are always hetero- geneous--not in bulk solution; therefore rate of reaction is usually normalized to surface are of electrode: V = mol/sec-cm2 = i/nFA = J/nF J= current density = i/A = amps/cm2 i-E curves used to obtain information about faradaic reaction. Departure of electrochemical cell from its equilibrium potential--Eeq--upon passage of a faradaic current is termed polarization. The extent of polarization is measured by the overpotential for the cell --- = E - Eeq Current-Voltage curves for electrochemical cells obtained under steady-state conditions are often termed polarization curves IPEs-show very large change in potential for passage of little or no faradaic current. A depolarizer is an electroactive species that causes the potential of an electrode to be nearer its equilib. value by being oxidized or reduced at the surface of the electrode. An ideally depolarized (or nonpolarizable) electrode is one whose potential does not change upon passage of faradaic current. i i IPE IDE E Eappl appl A saturated calomel electrode---prepared with a large surface area mercury pool---would represent and IDE------See figure below: wire glass KCl soln tube Hg2Cl2(s) Hg If you have large surface area for reference elect. -

Introduction
CHAPTER 1 INTRODUCTION Electrochemical techniques are used for the production of aluminum and chlorine, the conversion of energy in batteries and fuel cells, sensors, electroplating, and the protection of metal structures against corrosion, to name just a few prominent applications. While applications such as fuel cells and electroplating may seem quite disparate, in this book we show that a few basic principles form the foundation for the design of all electrochemical processes. The first practical electrochemical system was the Volta pile, invented by Alexander Volta in 1800. Volta’s pile is still used today in batteries for a variety of industrial, medical, and military applications. Volta found that when he made a sandwich of a layer of zinc metal, paper soaked in salt water, and tarnished silver and then connected a wire from the zinc to the silver, he could obtain electricity (see Figure 1.1). What is happening when the wire is connected? Electrons have a chemical preference to be in the silver rather than the zinc, and this chemical preference is manifest as a voltage difference that drives the current. At each electrode, an electrochemical reaction is occurring: zinc reacts with hydroxide ions in solution to form free electrons, zinc oxide, and water, while silver oxide (tarnished silver) reacts with water and electrons to form silver and hydroxide ions. Hydroxide ions travel through the salt solution (the electrolyte) from the silver to the zinc, while electrons travel through the external wire from the zinc to the silver. We see from this example that many phenomena interact in electrochemical systems.