Integrated Transcriptomic and Neuroimaging Brain Model Decodes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Datasheet Blank Template
SAN TA C RUZ BI OTEC HNOL OG Y, INC . Fe65L (Y-14): sc-104237 BACKGROUND PRODUCT Fe65L (Fe65-like protein), also known as APBB2 (amyloid β (A4) precursor Each vial contains 200 µg IgG in 1.0 ml of PBS with < 0.1% sodium azide protein-binding, family B, member 2), is a 758 amino acid protein that contains and 0.1% gelatin. one WW domain and 2 PID domains. Binding to the intracellular domain of Blocking peptide available for competition studies, sc-104237 P, (100 µg the -amyloid precursor protein, Fe65L is thought to modulate the internal - β peptide in 0.5 ml PBS containing < 0.1% sodium azide and 0.2% BSA). ization and, therefore, the accessibility and function of β-amyloid. Via its ability to control the intracellular accumulation of -amyloid, Fe65L is thought β APPLICATIONS to play a role in the pathogenesis of Alzheimer’s disease. Multiple isoforms of Fe65L exist due to alternative splicing events. The gene encoding Fe65L Fe65L (Y-14) is recommended for detection of Fe65L of mouse, rat and maps to human chromosome 4, which encodes nearly 6% of the human human origin by Western Blotting (starting dilution 1:200, dilution range genome and has the largest gene deserts (regions of the genome with no 1:100-1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein protein encoding genes) of all of the human chromosomes. Defects in some (1 ml of cell lysate)], immunofluorescence (starting dilution 1:50, dilution of the genes located on chromosome 4 are associated with Huntington’s range 1:50-1:500) and solid phase ELISA (starting dilution 1:30, dilution dis ease, Ellis-van Creveld syndrome, methylmalonic acidemia and polycystic range 1:30-1:3000). -
Final Version
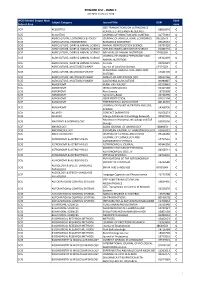
REWARD LIST ‐ RANK C JCR 2018 ‐CiteScore 2018 WOS Edition/ Scopus Main Rank Subject Category Journal Title ISSN Subject Area code IEEE TRANSACTIONS ON ULTRASONICS SCIE ACOUSTICS 08853010 C FERROELECTRICS AND FREQUENCY SCIE ACOUSTICS JOURNAL OF VIBRATION AND CONTROL 10775463 C SCIE AGRICULTURAL ECONOMICS & POLICY JOURNAL OF AGRICULTURAL ECONOMICS 0021857X C SCIE AGRICULTURAL ENGINEERING BIOMASS & BIOENERGY 09619534 C SCIE AGRICULTURE, DAIRY & ANIMAL SCIENCE ANIMAL REPRODUCTION SCIENCE 03784320 C SCIE AGRICULTURE, DAIRY & ANIMAL SCIENCE APPLIED ANIMAL BEHAVIOUR SCIENCE 01681591 C SCIE AGRICULTURE, DAIRY & ANIMAL SCIENCE ARCHIVES OF ANIMAL NUTRITION 1745039X C JOURNAL OF ANIMAL PHYSIOLOGY AND SCIE AGRICULTURE, DAIRY & ANIMAL SCIENCE 09312439 C ANIMAL NUTRITION SCIE AGRICULTURE, DAIRY & ANIMAL SCIENCE Animals 20762615 C SCIE AGRICULTURE, MULTIDISCIPLINARY Journal of Land Use Science 1747423X C RENEWABLE AGRICULTURE AND FOOD SCIE AGRICULTURE, MULTIDISCIPLINARY 17421705 C SYSTEMS SCIE AGRICULTURE, MULTIDISCIPLINARY ANNALS OF APPLIED BIOLOGY 00034746 C SCIE AGRICULTURE, MULTIDISCIPLINARY CALIFORNIA AGRICULTURE 00080845 C SCIE AGRONOMY PLANT PATHOLOGY 00320862 C SCIE AGRONOMY IRRIGATION SCIENCE 03427188 C SCIE AGRONOMY Rice Science 16726308 C SCIE AGRONOMY Agronomy‐Basel 20734395 C SCIE AGRONOMY CROP PROTECTION 02612194 C SCIE AGRONOMY EXPERIMENTAL AGRICULTURE 00144797 C JOURNAL OF PLANT NUTRITION AND SOIL SCIE AGRONOMY 14368730 C SCIENCE SCIE ALLERGY CONTACT DERMATITIS 01051873 C SCIE ALLERGY Allergy Asthma & Immunology Research 20927355 C Advances -

Identification of Eqtls and Sqtls Associated with Meat Quality in Beef Joel D
Leal-Gutiérrez et al. BMC Genomics (2020) 21:104 https://doi.org/10.1186/s12864-020-6520-5 RESEARCH ARTICLE Open Access Identification of eQTLs and sQTLs associated with meat quality in beef Joel D. Leal-Gutiérrez*, Mauricio A. Elzo and Raluca G. Mateescu Abstract Background: Transcription has a substantial genetic control and genetic dissection of gene expression could help us understand the genetic architecture of complex phenotypes such as meat quality in cattle. The objectives of the present research were: 1) to perform eQTL and sQTL mapping analyses for meat quality traits in longissimus dorsi muscle; 2) to uncover genes whose expression is influenced by local or distant genetic variation; 3) to identify expression and splicing hot spots; and 4) to uncover genomic regions affecting the expression of multiple genes. Results: Eighty steers were selected for phenotyping, genotyping and RNA-seq evaluation. A panel of traits related to meat quality was recorded in longissimus dorsi muscle. Information on 112,042 SNPs and expression data on 8588 autosomal genes and 87,770 exons from 8467 genes were included in an expression and splicing quantitative trait loci (QTL) mapping (eQTL and sQTL, respectively). A gene, exon and isoform differential expression analysis previously carried out in this population identified 1352 genes, referred to as DEG, as explaining part of the variability associated with meat quality traits. The eQTL and sQTL mapping was performed using a linear regression model in the R package Matrix eQTL. Genotype and year of birth were included as fixed effects, and population structure was accounted for by including as a covariate the first PC from a PCA analysis on genotypic data. -

Downloaded the “Top Edge” Version
bioRxiv preprint doi: https://doi.org/10.1101/855338; this version posted December 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Drosophila models of pathogenic copy-number variant genes show global and 2 non-neuronal defects during development 3 Short title: Non-neuronal defects of fly homologs of CNV genes 4 Tanzeen Yusuff1,4, Matthew Jensen1,4, Sneha Yennawar1,4, Lucilla Pizzo1, Siddharth 5 Karthikeyan1, Dagny J. Gould1, Avik Sarker1, Yurika Matsui1,2, Janani Iyer1, Zhi-Chun Lai1,2, 6 and Santhosh Girirajan1,3* 7 8 1. Department of Biochemistry and Molecular Biology, Pennsylvania State University, 9 University Park, PA 16802 10 2. Department of Biology, Pennsylvania State University, University Park, PA 16802 11 3. Department of Anthropology, Pennsylvania State University, University Park, PA 16802 12 4 contributed equally to work 13 14 *Correspondence: 15 Santhosh Girirajan, MBBS, PhD 16 205A Life Sciences Building 17 Pennsylvania State University 18 University Park, PA 16802 19 E-mail: [email protected] 20 Phone: 814-865-0674 21 1 bioRxiv preprint doi: https://doi.org/10.1101/855338; this version posted December 6, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 22 ABSTRACT 23 While rare pathogenic copy-number variants (CNVs) are associated with both neuronal and non- 24 neuronal phenotypes, functional studies evaluating these regions have focused on the molecular 25 basis of neuronal defects. -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -

Phosphatidylglycerol Incorporates Into Cardiolipin to Improve
www.nature.com/scientificreports OPEN Phosphatidylglycerol Incorporates into Cardiolipin to Improve Mitochondrial Activity and Inhibits Received: 3 July 2017 Accepted: 7 March 2018 Infammation Published: xx xx xxxx Wei-Wei Chen1, Yu-Jen Chao1, Wan-Hsin Chang1, Jui-Fen Chan1 & Yuan-Hao Howard Hsu1,2 Chronic infammation and concomitant oxidative stress can induce mitochondrial dysfunction due to cardiolipin (CL) abnormalities in the mitochondrial inner membrane. To examine the responses of mitochondria to infammation, macrophage-like RAW264.7 cells were activated by Kdo2-Lipid A (KLA) in our infammation model, and then the mitochondrial CL profle, mitochondrial activity, and the mRNA expression of CL metabolism-related genes were examined. The results demonstrated that KLA activation caused CL desaturation and the partial loss of mitochondrial activity. KLA activation also induced the gene upregulation of cyclooxygenase (COX)-2 and phospholipid scramblase 3, and the gene downregulation of COX-1, lipoxygenase 5, and Δ-6 desaturase. We further examined the phophatidylglycerol (PG) inhibition efects on infammation. PG supplementation resulted in a 358- fold inhibition of COX-2 mRNA expression. PG(18:1)2 and PG(18:2)2 were incorporated into CLs to considerably alter the CL profle. The decreased CL and increased monolysocardiolipin (MLCL) quantity resulted in a reduced CL/MLCL ratio. KLA-activated macrophages responded diferentially to PG(18:1)2 and PG(18:2)2 supplementation. Specifcally, PG(18:1)2 induced less changes in the CL/MLCL ratio than did PG(18:2)2, which resulted in a 50% reduction in the CL/MLCL ratio. However, both PG types rescued 20–30% of the mitochondrial activity that had been afected by KLA activation. -

Role and Regulation of Snon/Skil and PLSCR1 Located at 3Q26.2
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School 9-18-2014 Role and Regulation of SnoN/SkiL and PLSCR1 Located at 3q26.2 and 3q23, Respectively, in Ovarian Cancer Pathophysiology Madhav Karthik Kodigepalli University of South Florida, [email protected] Follow this and additional works at: https://scholarcommons.usf.edu/etd Part of the Cell Biology Commons, Microbiology Commons, and the Molecular Biology Commons Scholar Commons Citation Kodigepalli, Madhav Karthik, "Role and Regulation of SnoN/SkiL and PLSCR1 Located at 3q26.2 and 3q23, Respectively, in Ovarian Cancer Pathophysiology" (2014). Graduate Theses and Dissertations. https://scholarcommons.usf.edu/etd/5426 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Role and Regulation of SnoN/SkiL and PLSCR1 Located at 3q26.2 and 3q23, Respectively, in Ovarian Cancer Pathophysiology by Madhav Karthik Kodigepalli A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Cell and Molecular Biology Department of Cell Biology, Microbiology and Molecular Biology College of Arts and Sciences University of South Florida Major Professor: Meera Nanjundan, Ph.D. Richard Pollenz, Ph.D. Patrick Bradshaw, Ph.D. Sandy Westerheide, Ph.D. Date of Approval: September 18, 2014 Keywords: Chemotherapeutics, phospholipid scramblase, toll-like receptor, interferon, dsDNA Copyright © 2014, Madhav Karthik Kodigepalli Dedication I dedicate this research at the lotus feet of Bhagwan Sri Sathya Sai Baba and all the Masters for I am what I am due to their divine grace. -

SCIENCE CITATION INDEX EXPANDED - JOURNAL LIST Total Journals: 8631
SCIENCE CITATION INDEX EXPANDED - JOURNAL LIST Total journals: 8631 1. 4OR-A QUARTERLY JOURNAL OF OPERATIONS RESEARCH 2. AAPG BULLETIN 3. AAPS JOURNAL 4. AAPS PHARMSCITECH 5. AATCC REVIEW 6. ABDOMINAL IMAGING 7. ABHANDLUNGEN AUS DEM MATHEMATISCHEN SEMINAR DER UNIVERSITAT HAMBURG 8. ABSTRACT AND APPLIED ANALYSIS 9. ABSTRACTS OF PAPERS OF THE AMERICAN CHEMICAL SOCIETY 10. ACADEMIC EMERGENCY MEDICINE 11. ACADEMIC MEDICINE 12. ACADEMIC PEDIATRICS 13. ACADEMIC RADIOLOGY 14. ACCOUNTABILITY IN RESEARCH-POLICIES AND QUALITY ASSURANCE 15. ACCOUNTS OF CHEMICAL RESEARCH 16. ACCREDITATION AND QUALITY ASSURANCE 17. ACI MATERIALS JOURNAL 18. ACI STRUCTURAL JOURNAL 19. ACM COMPUTING SURVEYS 20. ACM JOURNAL ON EMERGING TECHNOLOGIES IN COMPUTING SYSTEMS 21. ACM SIGCOMM COMPUTER COMMUNICATION REVIEW 22. ACM SIGPLAN NOTICES 23. ACM TRANSACTIONS ON ALGORITHMS 24. ACM TRANSACTIONS ON APPLIED PERCEPTION 25. ACM TRANSACTIONS ON ARCHITECTURE AND CODE OPTIMIZATION 26. ACM TRANSACTIONS ON AUTONOMOUS AND ADAPTIVE SYSTEMS 27. ACM TRANSACTIONS ON COMPUTATIONAL LOGIC 28. ACM TRANSACTIONS ON COMPUTER SYSTEMS 29. ACM TRANSACTIONS ON COMPUTER-HUMAN INTERACTION 30. ACM TRANSACTIONS ON DATABASE SYSTEMS 31. ACM TRANSACTIONS ON DESIGN AUTOMATION OF ELECTRONIC SYSTEMS 32. ACM TRANSACTIONS ON EMBEDDED COMPUTING SYSTEMS 33. ACM TRANSACTIONS ON GRAPHICS 34. ACM TRANSACTIONS ON INFORMATION AND SYSTEM SECURITY 35. ACM TRANSACTIONS ON INFORMATION SYSTEMS 36. ACM TRANSACTIONS ON INTELLIGENT SYSTEMS AND TECHNOLOGY 37. ACM TRANSACTIONS ON INTERNET TECHNOLOGY 38. ACM TRANSACTIONS ON KNOWLEDGE DISCOVERY FROM DATA 39. ACM TRANSACTIONS ON MATHEMATICAL SOFTWARE 40. ACM TRANSACTIONS ON MODELING AND COMPUTER SIMULATION 41. ACM TRANSACTIONS ON MULTIMEDIA COMPUTING COMMUNICATIONS AND APPLICATIONS 42. ACM TRANSACTIONS ON PROGRAMMING LANGUAGES AND SYSTEMS 43. ACM TRANSACTIONS ON RECONFIGURABLE TECHNOLOGY AND SYSTEMS 44. -

Viewed and Published Immediately Upon Acceptance Cited in Pubmed and Archived on Pubmed Central Yours — You Keep the Copyright
BMC Genomics BioMed Central Research article Open Access Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice Claudia Prinzen1, Dietrich Trümbach2, Wolfgang Wurst2, Kristina Endres1, Rolf Postina1 and Falk Fahrenholz*1 Address: 1Johannes Gutenberg-University, Institute of Biochemistry, Mainz, Johann-Joachim-Becherweg 30, 55128 Mainz, Germany and 2Helmholtz Zentrum München – German Research Center for Environmental Health, Institute for Developmental Genetics, Ingolstädter Landstraße 1, 85764 Neuherberg, Germany Email: Claudia Prinzen - [email protected]; Dietrich Trümbach - [email protected]; Wolfgang Wurst - [email protected]; Kristina Endres - [email protected]; Rolf Postina - [email protected]; Falk Fahrenholz* - [email protected] * Corresponding author Published: 5 February 2009 Received: 19 June 2008 Accepted: 5 February 2009 BMC Genomics 2009, 10:66 doi:10.1186/1471-2164-10-66 This article is available from: http://www.biomedcentral.com/1471-2164/10/66 © 2009 Prinzen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: In a transgenic mouse model of Alzheimer disease (AD), cleavage of the amyloid precursor protein (APP) by the α-secretase ADAM10 prevented amyloid plaque formation, and alleviated cognitive deficits. Furthermore, ADAM10 overexpression increased the cortical synaptogenesis. These results suggest that upregulation of ADAM10 in the brain has beneficial effects on AD pathology. Results: To assess the influence of ADAM10 on the gene expression profile in the brain, we performed a microarray analysis using RNA isolated from brains of five months old mice overexpressing either the α-secretase ADAM10, or a dominant-negative mutant (dn) of this enzyme. -

Jennifer I. Luebke 1 Curriculum Vitae Jennifer I. Luebke, Ph.D. Associate
Jennifer I. Luebke Curriculum Vitae Jennifer I. Luebke, Ph.D. Associate Professor of Anatomy & Neurobiology Associate Professor of Psychiatry Director, Laboratory of Cellular Neurobiology Boston University School of Medicine 85 East Newton Street, M949 Boston, Massachusetts 02118 Voice: 617-638-4930 Email: [email protected] Education and Employment History: 1980-1984: B.S. (Biology) Randolph-Macon College, Ashland, Virginia 1984-1986: Laboratory Technician (Laboratory of Cell Biology and Genetics, NIDDK) National Institutes of Health, Bethesda, Maryland 1986-1990: Ph.D. Student (Anatomy & Neurobiology), Boston University School of Medicine, Boston, Massachusetts (Linda L. Wright, mentor) 1990-1992: Postdoctoral Fellow, Department of Psychiatry, Harvard Medical School, Boston, Massachusetts (Robert W. McCarley and Robert W. Greene mentors) 1992-1995: Postdoctoral Fellow, Department of Physiology, Tufts University Medical School, Boston, Massachusetts (Kathleen Dunlap, mentor) 1995-2003: Assistant Professor, Department of Anatomy & Neurobiology and Department of Psychiatry, Boston University School of Medicine, Boston, Massachusetts 2004-Present: Associate Professor, Department of Anatomy & Neurobiology and Department of Psychiatry, Boston University School of Medicine, Boston, Massachusetts 2010-Present: Adjunct Associate Professor, Department of Neuroscience, Mount Sinai School of Medicine, New York, New York Research Summary: Research is directed toward understanding alterations in the structure and function of individual cortical pyramidal cells in a rhesus monkey model of normal aging and in transgenic mouse models of neurodegenerative disease. Using whole-cell patch-clamp methods and ultra-high resolution confocal microscopy, Dr. Luebke has demonstrated marked alterations in action potential firing patterns (and underlying ionic currents), glutamatergic and GABAergic synaptic response properties and detailed dendritic and spine architecture in cortical pyramidal cells both in normal aging and in mouse models of neurodegenerative disease. -

Predicting Cognitive Decline: Genetic, Environmental and Lifestyle Risk Factors
Predicting Cognitive Decline: Genetic, Environmental and Lifestyle Risk Factors Shea J. Andrews April 2017 A thesis by compilation submitted for the degree of Doctor of Philosophy of The Australian National University © Copyright by Shea John Frederick Andrews 2017 All Rights Reserved i Declaration This work was conducted from February 2013 to August 2016 at the Genome Diversity and Health Group, John Curtin School of Medical Research, The Australian National University, Canberra, ACT. This thesis by compilation consists of five original publications describing the analyses I have performed during my candidature investigating the role of genetic, environmental and lifestyle risk factors in normal cognitive aging. All five publications have been published in Q1 ranked journals according to the SCImago Journal & Country Rankings in the fields of neurology, genetics, psychiatry and mental health, and geriatric and gerontology. My specific contribution to each manuscript is detailed in the subsequent pages in the form of a statement signed by the senior author of each publication. This document has not been submitted for qualifications at any other academic institution. Shea Andrews Canberra, Australia April 2017 ii Published Papers Andrews, SJ, Das, D., Anstey, KJ., Easteal, S. (2015). Interactive effect of APOE genotype and blood pressure on cognitive decline: The PATH through life project. Journal of Alzheimer's Disease. 44(4): 1087-98. DOI: 10.3233/JAD- 140630 For this publication, I designed and performed all statistical analyses and wrote the manuscript. A slightly modified version of this paper is presented in Chapter 3. ____________________ Simon Easteal Senior Author April 2017 Andrews SJ, Das D, Cherbuin N, Anstey KJ, Easteal S. -

Effects of Engineered Costimulation on the Function of T Cell Subsets
Effects of Engineered Costimulation on the Function of T Cell Subsets The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Boroughs, Angela C. 2019. Effects of Engineered Costimulation on the Function of T Cell Subsets. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:41121316 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Effects of Engineered Costimulation on the Function of T Cell Subsets A dissertation presented by Angela Clare Boroughs to The Division of Medical Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Immunology Harvard University Cambridge, Massachusetts October 2018 © 2018 Angela Clare Boroughs All rights reserved. Dissertation Advisor: Dr. Marcela V. Maus Angela Clare Boroughs Effects of Engineered Costimulation on the Function of T Cell Subsets Abstract Progress in clinical adoptive immunotherapy was initially hindered by the cells’ lack of antigen specificity, poor engraftment, and limited persistence in the host. The introduction of chimeric antigen receptors (CARs) into T cells has overcome these obstacles by re-directing polyclonal T cells to a specific antigen with an extracellular binding domain and by including costimulation domains that enhance engraftment and persistence. From this observation, we hypothesized that the overexpression of CARs in different T cell subsets fundamentally changes their biology.