Supplementary Appendix Integrated Genomic Analysis of Recurrence-Associated Small Non-Coding Rnas in Oesophageal Cancer Hee-Jin
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Topological Scoring of Protein Interaction Networks
bioRxiv preprint doi: https://doi.org/10.1101/438408; this version posted October 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Topological Scoring of Protein Interaction Networks Mihaela E. Sardiu1, Joshua M. Gilmore1,2, Brad D. Groppe1,3, Arnob Dutta1,4, Laurence Florens1, and Michael P. Washburn1,5‡ 1Stowers Institute for Medical Research, Kansas City, MO 64110 U.S.A. 2Current Address: Boehringer Ingelheim Vetmedica, St. Joseph, MO 64506 U.S.A. 3Current Address: Thermo Fisher Scientific, Waltham, MA 02451, U.S.A. 4Current Address: Department of Cell and Molecular Biology, University of Rhode Island, 287 CBLS, 120 Flagg Road, Kingston, RI 02881. 5 Department of Pathology and Laboratory Medicine, The University of Kansas Medical Center, 3901 Rainbow Boulevard, Kansas City, Kansas 66160, USA ‡To whom correspondence should be addressed: Michael Washburn, Ph.D. Stowers Institute for Medical Research 1000 E. 50th St. Kansas City, MO 64110 Phone: 816-926-4457 E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/438408; this version posted October 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract It remains a significant challenge to define individual protein associations within networks where an individual protein can directly interact with other proteins and/or be part of large complexes, which contain functional modules. Here we demonstrate the topological scoring (TopS) algorithm for the analysis of quantitative proteomic analyses of affinity purifications. -

Genetic Causes of Severe Childhood Obesity: a Remarkably High Prevalence in an Inbred Population of Pakistan
1424 Diabetes Volume 69, July 2020 Genetic Causes of Severe Childhood Obesity: A Remarkably High Prevalence in an Inbred Population of Pakistan Sadia Saeed,1,2 Muhammad Arslan,3 Jaida Manzoor,4 Sadia M. Din,5 Qasim M. Janjua,5,6 Hina Ayesha,7 Qura-tul Ain,5 Laraib Inam,3 Stephane Lobbens,1 Emmanuel Vaillant,1 Emmanuelle Durand,1 Mehdi Derhourhi,1 Souhila Amanzougarene,1 Alaa Badreddine,1 Lionel Berberian,1 Stefan Gaget,1 Waqas I. Khan,8 Taeed A. Butt,9 Amélie Bonnefond,1,2 and Philippe Froguel1,2 Diabetes 2020;69:1424–1438 | https://doi.org/10.2337/db19-1238 Monogenic forms of obesity have been identified in £10% material in the quest of new genes/variants influencing of severely obese European patients. However, the overall energy balance. spectrum of deleterious variants (point mutations and structural variants) responsible for childhood severe obe- sity remains elusive. In this study, we genetically screened The monogenic forms of obesity have defined the current 225 severely obese children from consanguineous Pakis- concepts of the central regulation of energy balance and have tani families through a combination of techniques, includ- opened new avenues for precision medicine (1,2). Monogenic ing an in-house–developed augmented whole-exome nonsyndromic obesity is due to pathogenic mutations in sequencing method (CoDE-seq) that enables simultaneous genes involved in leptin-melanocortin signaling, resulting in detection of whole-exome copy number variations (CNVs) extreme, early-onset obesity with an insatiable craving for fi and point mutations in coding regions. We identi ed food (2). In addition to excessive adiposity, syndromic obesity OBESITY STUDIES 110 (49%) probands carrying 55 different pathogenic point associates with other abnormalities such as dysmorphic fea- mutations and CNVs in 13 genes/loci responsible for non- tures, intellectual disability, and organ-specific anomalies (3). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Distinguishing Pleiotropy from Linked QTL Between Milk Production Traits
Cai et al. Genet Sel Evol (2020) 52:19 https://doi.org/10.1186/s12711-020-00538-6 Genetics Selection Evolution RESEARCH ARTICLE Open Access Distinguishing pleiotropy from linked QTL between milk production traits and mastitis resistance in Nordic Holstein cattle Zexi Cai1*†, Magdalena Dusza2†, Bernt Guldbrandtsen1, Mogens Sandø Lund1 and Goutam Sahana1 Abstract Background: Production and health traits are central in cattle breeding. Advances in next-generation sequencing technologies and genotype imputation have increased the resolution of gene mapping based on genome-wide association studies (GWAS). Thus, numerous candidate genes that afect milk yield, milk composition, and mastitis resistance in dairy cattle are reported in the literature. Efect-bearing variants often afect multiple traits. Because the detection of overlapping quantitative trait loci (QTL) regions from single-trait GWAS is too inaccurate and subjective, multi-trait analysis is a better approach to detect pleiotropic efects of variants in candidate genes. However, large sample sizes are required to achieve sufcient power. Multi-trait meta-analysis is one approach to deal with this prob- lem. Thus, we performed two multi-trait meta-analyses, one for three milk production traits (milk yield, protein yield and fat yield), and one for milk yield and mastitis resistance. Results: For highly correlated traits, the power to detect pleiotropy was increased by multi-trait meta-analysis com- pared with the subjective assessment of overlapping of single-trait QTL confdence intervals. Pleiotropic efects of lead single nucleotide polymorphisms (SNPs) that were detected from the multi-trait meta-analysis were confrmed by bivariate association analysis. The previously reported pleiotropic efects of variants within the DGAT1 and MGST1 genes on three milk production traits, and pleiotropic efects of variants in GHR on milk yield and fat yield were con- frmed. -

The Evolutionarily Conserved Kruppel-Associated Box Domain
Proc. Natl. Acad. Sci. USA Vol. 88, pp. 3608-3612, May 1991 Biochemistry The evolutionarily conserved Kruppel-associated box domain defines a subfamily of eukaryotic multifingered proteins (DNA-binding proteins/sequence conservation/ceil differentiation) ERIC J. BELLEFROID, DOMINIQUE A. PONCELET, PIERRE J. LECOCQ, OLIVIER REVELANT, AND JOSEPH A. MARTIAL Laboratoire de Biologie Moldculaire et de Genie G6ndtique, Universit6 de Liege, Institut de Chimie B6, B-4000 Sart Tilman, Belgium Communicated by William J. Rutter, January 2, 1991 ABSTRACT We have previously shown that the human number of finger proteins (20-23), is highly conserved in genome includes hundreds of genes coding for putative factors evolution and appears always associated with finger repeats. related to the Krfippel zinc-ringer protein, which regulates Drosophila segmentation. We report herein that about one- third of these genes code for proteins that share a very MATERIALS AND METHODS conserved region of about 75 amino acids in their N-terminal cDNA Library Screening. A human AgtlO cDNA library nonfinger portion. Homologous regions are found in a number from undifferentiated NT2D1 cells (24) was screened with a of previously described finger proteins, including mouse Zfp-l 546-base-pair EcoRI-EcoRI fragment corresponding to the 5' and Xenopus Xfin. We named this region the Kruppel- nonfinger portion of the coding region of HPF4 (where HPF associated box (KRAB). This domain has the potential to form is human placental finger) (19). The DNA probes were two amphipathic a-helices. Southern blot analysis of "zoo" labeled with a multiprimed labeling kit (Boehringer Mann- blots suggests that the Krfippel-associated box is highly con- heim) to a specific activity of5 x 101 cpm/pug. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Characterization and Mapping of Human Genes Encoding Zinc Finger Proteins (Transcription/Chromosome/Sequence-Tagged Site) P
Proc. Nati. Acad. Sci. USA Vol. 88, pp. 9563-9567, November 1991 Biochemistry Characterization and mapping of human genes encoding zinc finger proteins (transcription/chromosome/sequence-tagged site) P. BRAY*, P. LICHTERtt, H.-J. THIESEN§, D. C. WARDt, AND I. B. DAWID* *Laboratory of Molecular Genetics, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892; tDepartment of Genetics, Yale School of Medicine, New Haven, CT 06520; *Deutsches Krebsforschungszentrum, Im Neuenheimer Feld 280, D-6900 Heidelberg, Federal Republic of Germany; §Basel Institute for Immunology, Grenzacherstrasse 487, CH-4005 Basel, Switzerland Contributed by I. B. Dawid, August 2, 1991 ABSTRACT The zinc finger motif, exemplified by a segment finger genes cloned by binding of known regulatory nucleo- of the Drosophila gap gene Krfippel, is a nudeic add-binding tide sequences, zinc finger clones isolated by sequence domain present in many transcription factors. To investigate the homology often contain many fingers and highly conserved gene family encoding this motifin the human genome, a placental H/C link regions. Among multifinger cDNAs at least two genomic library was screened at moderate stringency with a associated motifs of unknown function have been identified degenerate oligodeoxynucleotide probe designed to hybridize to and named FAX (finger-associated box; ref. 14) and KRAB the His/Cys (H/C) link region between adjoining zinc fingers. (Kirppel-associated box; ref. 15). Over 200 phage clones were obtained and are being sorted into With the aim to survey the number and chromosomal groups by partial sequencing, cross-hybridization with oligode- distribution of zinc finger genes in the human genome, we oxynucleotide probes, and PCR amplifion. -

Whole Exome Sequencing Reveals NOTCH1 Mutations in Anaplastic Large Cell Lymphoma and Points to Notch Both As a Key Pathway and a Potential Therapeutic Target
Non-Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing reveals NOTCH1 mutations in anaplastic large cell lymphoma and points to Notch both as a key pathway and a potential therapeutic target Hugo Larose, 1,2 Nina Prokoph, 1,2 Jamie D. Matthews, 1 Michaela Schlederer, 3 Sandra Högler, 4 Ali F. Alsulami, 5 Stephen P. Ducray, 1,2 Edem Nuglozeh, 6 Mohammad Feroze Fazaludeen, 7 Ahmed Elmouna, 6 Monica Ceccon, 2,8 Luca Mologni, 2,8 Carlo Gambacorti-Passerini, 2,8 Gerald Hoefler, 9 Cosimo Lobello, 2,10 Sarka Pospisilova, 2,10,11 Andrea Janikova, 2,11 Wilhelm Woessmann, 2,12 Christine Damm-Welk, 2,12 Martin Zimmermann, 13 Alina Fedorova, 14 Andrea Malone, 15 Owen Smith, 15 Mariusz Wasik, 2,16 Giorgio Inghirami, 17 Laurence Lamant, 18 Tom L. Blundell, 5 Wolfram Klapper, 19 Olaf Merkel, 2,3 G. A. Amos Burke, 20 Shahid Mian, 6 Ibraheem Ashankyty, 21 Lukas Kenner 2,3,22 and Suzanne D. Turner 1,2,10 1Department of Pathology, University of Cambridge, Cambridge, UK; 2European Research Initiative for ALK Related Malignancies (ERIA; www.ERIALCL.net ); 3Department of Pathology, Medical University of Vienna, Vienna, Austria; 4Unit of Laboratory Animal Pathology, Uni - versity of Veterinary Medicine Vienna, Vienna, Austria; 5Department of Biochemistry, University of Cambridge, Tennis Court Road, Cam - bridge, UK; 6Molecular Diagnostics and Personalised Therapeutics Unit, Colleges of Medicine and Applied Medical Sciences, University of Ha’il, Ha’il, Saudi Arabia; 7Neuroinflammation Research Group, Department of Neurobiology, A.I Virtanen Institute -
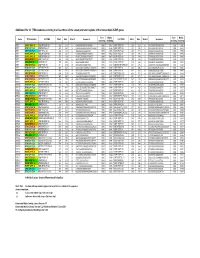
Additional File 14: TFBS Modules Occuring in at Least Three of the Seven Promoter Regions of the Human 8Q24.3 ZNF Genes
Additional file 14: TFBS modules occuring in at least three of the seven promoter regions of the human 8q24.3 ZNF genes Core Matrix Core Matrix Gene TFBS module 1st TFBS Start End Strand Sequence 2nd TFBS Start End Strand Sequence similarity similarity similarity similarity ZNF7 EGRF_SP1F_01 V$EGRF/EGR1.02 262 278 (-) CAGGGGCGGGGCGGGGC 1.000 0.907 V$SP1F/SP1.01 259 273 (-) GCGGGGCGGGGCCGG 1.000 1.000 ZNF7 ETSF_SP1F_03 V$ETSF/PEA3.01 301 321 (-) GAGGGAGAGGAGGGCCCAGAG 1.000 0.836 V$SP1F/SP1.03 281 295 (-) AGTGGGCGGTGCCTG 1.000 0.932 ZNF7 SP1F_AP2F_01 V$SP1F/GC.01 322 336 (-) AGGGGGCGGAGCCGC 1.000 0.965 V$AP2F/AP2.02 297 311 (-) AGGGCCCAGAGCCCG 0.905 0.795 ZNF7 EGRF_SP1F_01 V$EGRF/EGR1.02 325 341 (-) CGGGCAGGGGGCGGAGC 1.000 0.924 V$SP1F/GC.01 322 336 (-) AGGGGGCGGAGCCGC 1.000 0.965 ZNF7 EGRF_SP1F_01 V$EGRF/EGR1.02 353 369 (-) GCGCCTGGGGGCGGGGT 1.000 0.903 V$SP1F/SP1.01 350 364 (-) TGGGGGCGGGGTACA 1.000 0.997 ZNF7 KLFS_SP1F_01 V$KLFS/KLF6.01 364 380 (-) AGGCGGGGCTGGCGCCT 1.000 0.932 V$SP1F/SP1.01 350 364 (-) TGGGGGCGGGGTACA 1.000 0.997 ZNF7 IKRS_AP2F_01 V$IKRS/IK2.01 390 402 (-) GGGCGGGACCAGA 1.000 0.919 V$AP2F/AP2.02 410 424 (-) GGGGCCCGAGGGACA 0.905 0.910 ZNF7 SP1F_ETSF_04 V$SP1F/GC.01 471 485 (+) GGGAGGCGGGGCTGA 1.000 0.930 V$ETSF/CETS1P54.01 451 471 (-) CAGGCTCCGGAGAAGGGGTGG 1.000 0.843 ZNF7 ETSF_SP1F_03 V$ETSF/ELK1.02 521 541 (+) GGGCGGCCGGAAGTTTGCGGG 1.000 0.980 V$SP1F/SP1.03 542 556 (+) GCGGGGCGGACGCGG 1.000 0.924 ZNF16 ETSF_SP1F_05 V$ETSF/ELK1.02 491 511 (+) GGCGGGCCGGAATTCGGGGCG 1.000 0.961 V$SP1F/GC.01 487 501 (+) GAGCGGCGGGCCGGA -

Supp Data.Pdf
Supplementary Methods Verhaak sub-classification The Verhaak called tumor subtype was assigned as determined in the Verhaak et al. manuscript, for those TCGA tumors that were included at the time. TCGA data for expression (Affymetrix platform, level 1) and methylation (Infinium platform level 3) were downloaded from the TCGA portal on 09/29/2011. Affymetrix data were processed using R and Biocondutor with a RMA algorithm using quantile normalization and custom CDF. Level 3 methylation data has already been processed and converted to a beta-value. The genes that make up the signature for each of the 4 Verhaak groups (i.e. Proneural, Neural, Mesenchymal, and Classical) were downloaded. For each tumor, the averaged expression of the 4 genes signatures was calculated generating 4 ‘metagene’ scores, one for each subtype, (Colman et al.2010) for every tumor. Then each subtype metagene score was z-score corrected to allow comparison among the 4 metagenes. Finally, for each tumor the subtype with the highest metagene z-score among the four was used to assign subtype. Thus by this method, a tumor is assigned to the group to which it has the strongest expression signature. The shortcoming is that if a tumor has a ‘dual personality’ – for instance has strong expression of both classical and mesenchymal signature genes, there is some arbitrariness to the tumor’s assignment. Western Blot Analysis and cell fractionation Western blot analysis was performed using standard protocols. To determine protein expression we used the following antibodies: TAZ (Sigma and BD Biosciences), YAP (Santa Cruz Biotechnology), CD44 (Cell Signaling), FN1 (BD Biosciences), TEAD1, TEAD2, TEAD3, TEAD4 (Santa Cruz Biotechnology), MST1, p-MST1, MOB1, LATS1, LATS2, 14-3-3-, 1 ACTG2, (Cell Signaling), p-LATS1/2 (Abcam), Flag (Sigma Aldrich), Actin (Calbiochem), CAV2 (BD Biosciences), CTGF (Santa Cruz), RUNX2 (Sigma Aldrich), Cylin A, Cyclin E, Cyclin B1, p-cdk1, p-cdk4 (Cell Signaling Technologies). -

Supplemental Material For
SUPPLEMENTAL MATERIAL FOR Coexpression network based on natural variation in human gene expression reveals gene interactions and functions Renuka Nayak, Michael Kearns, Richard S. Spielman, Vivian G. Cheung Supplementary Figure 1 Supplementary Table 1. Gene pairs whose correlations in gene expression levels differ significantly (Pc<0.05) among the 3 datasets. Supplementary Table 2. Gene pairs that are correlated in gene expression levels with |R|>0.5 and are found within 500 kb of each other. Supplementary Table 3. Predicted functions of poorly characterized genes based on the functions of neighboring genes. Supplementary Figure 1. Genes identified in genome-wide association studies (grey) and their neighbors in the network. Red and green connections refer to positive and negative correlations, respectively. MICB has been implicated in AIDS progression (PMID: 19115949) TNF has been implicated in AIDS progression (PMID: 19115949) LTB has been implicated in AIDS progression (PMID: 19115949) ZNF224 has been implicated in Alzheimer's disease (PMID: 19118814) NDUFAB1 has been implicated in bipolar disorder (PMID: 17554300) SFRS10 has been implicated in body mass index (PMID: 19079260) and weight (PMID: 19079260) CTNNBL1 has been implicated in bone mineral density (PMID: 17903296) TGFBR3 has been implicated in bone mineral density (PMID: 19249006) IGF2R has been implicated in brain lesion load (PMID: 19010793) LSP1 has been implicated in breast cancer (PMID: 17529967) FBN1 has been implicated in breast cancer (PMID: 17903305) GLG1 has been implicated in breast cancer (PMID: 18463975) SCHIP1 has been implicated in Celiac disease (PMID: 18311140) RGS1 has been implicated in Celiac disease (PMID: 18311140) FADS2 has been implicated in Cholesterol (total) (PMID: 19060911), HDL cholesterol (PMID: 19060911, 19060906), LDL cholesterol (PMID: 19060911, 19060910), and triglycerides (PMID: 19060906). -

A Dynamic Model of Gene Expression in Monocytes Reveals Differences In
Journal of Inflammation BioMed Central Research Open Access A dynamic model of gene expression in monocytes reveals differences in immediate/early response genes between adult and neonatal cells Shelley Lawrence†1, Yuhong Tang†2, M Barton Frank2, Igor Dozmorov2, Kaiyu Jiang1, Yanmin Chen1, Craig Cadwell2, Sean Turner2, Michael Centola2 and James N Jarvis*1 Address: 1Dept. of Pediatrics, Neonatal Section, University of Oklahoma College of Medicine, Oklahoma City, OK, USA and 2Arthritis & Immunology Program Oklahoma Medical Research Foundation, Oklahoma City, 73104, USA Email: Shelley Lawrence - [email protected]; Yuhong Tang - [email protected]; M Barton Frank - Bart- [email protected]; Igor Dozmorov - [email protected]; Kaiyu Jiang - [email protected]; Yanmin Chen - yanmin- [email protected]; Craig Cadwell - [email protected]; Sean Turner - [email protected]; Michael Centola - michael- [email protected]; James N Jarvis* - [email protected] * Corresponding author †Equal contributors Published: 16 February 2007 Received: 26 September 2006 Accepted: 16 February 2007 Journal of Inflammation 2007, 4:4 doi:10.1186/1476-9255-4-4 This article is available from: http://www.journal-inflammation.com/content/4/1/4 © 2007 Lawrence et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Neonatal monocytes display immaturity of numerous functions compared with adult cells. Gene expression arrays provide a promising tool for elucidating mechanisms underlying neonatal immune function.