XU-DOCUMENT-2014.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Products of OH + Furan Reactions and Some Implications for Aromatic Hydrocarbon Atmospheric Degradation
Products of OH + Furan Reactions and Some Implications for Aromatic Hydrocarbon Atmospheric Degradation Roger Atkinson, Air Pollution Research Center, University of California, Riverside Unsaturated 1,4-dicarbonyls are important products of the atmospheric degradations of aromatic hydrocarbons such as benzene, toluene, xylenes and trimethylbenzenes, which comprise 20% of the non- methane volatile organic compounds present in urban air masses in the USA. However, in many cases the measured formation yields of the unsaturated 1,4-dicarbonyls are significantly lower than those of these presumed co-product 1,2-dicarbonyls. These discrepancies could be due to analytical problems and/or rapid photolysis of unsaturated 1,4-keto-aldehydes and unsaturated 1,4-dialdehydes, or to incorrect reaction mechanisms for the OH radical-initiated reactions of aromatic hydrocarbons. Since unsaturated 1,4- dicarbonyls are major products of OH + furans, with apparently simpler product distributions and mechanisms, we have investigated the reactions of OH radicals with furan, 2- and 3-methylfuran, and 2,3- and 2,5- dimethylfuran, in the presence of NO. Using direct air sampling atmospheric pressure ionization tandem mass spectrometry and gas chromatography, the unsaturated 1,4-dicarbonyls were observed and quantified. The measured unsaturated 1,4-dicarbonyl formation yields ranged from 8 ± 2% from OH + 2,3-dimethylfuran to 75 ± 5% from OH + furan. Other products were also formed. These data will be presented and discussed and, time permitting, a brief discussion of in situ nitro-aromatic and nitro-PAH formation from the atmospheric degradations of aromatic hydrocarbons and PAHs will also be presented. Live cast on the link below: http://www.fin.ucar.edu/it/mms/fl-live.htm A recording will be available on ACD’s website for viewing at a later date. -

Formation of Highly Oxygenated Organic Molecules from Aromatic Compounds
Formation of highly oxygenated organic molecules from aromatic compounds. Ugo Molteni1, Federico Bianchi1-2, Felix Klein1, Imad El Haddad1, Carla Frege1, Michel J. Rossi1, Josef Dommen1, Urs Baltensperger1,* 5 1Laboratory of Atmospheric Chemistry, Paul Scherrer Institute, CH-5232 Villigen, Switzerland 2Department of Physics, University of Helsinki, 00014 Helsinki, Finland Correspondence to: Urs Baltensperger ([email protected]) Abstract 10 Anthropogenic volatile organic compounds (AVOC) often dominate the urban atmosphere and consist to a large degree of aromatic hydrocarbons (ArHC), such as benzene, toluene, xylenes, and trimethylbenzenes, e.g. from handling and combustion of fuels. These compounds are important precursors for the formation of secondary organic aerosol. Despite their recognized importance as atmospheric reactants, the formation of highly oxygenated molecules (HOMs) in the gas phase leading to (extremely) low volatility compounds has not been studied in the past. Here we show that oxidation of 15 aromatics with OH leads to a subsequent autoxidation chain reaction forming HOMs with an O:C ratio of up to 1.09. This is exemplified for five single-ring ArHC (benzene, toluene, o-/m-/p-xylene, mesitylene (1,3,5-trimethylbenzene) and ethylbenzene), as well as two conjugated polycyclic ArHC (naphthalene and biphenyl). We present the identified compounds, differences in the observed oxidation patterns and discuss mechanistic pathways. We report the elemental composition of the HOMs and show the differences in the oxidation patterns of these ArHCs. A potential pathway for the 20 formation of these HOMs from aromatics is presented and discussed. We hypothesize that AVOC may contribute substantially to new particle formation events that have been detected in urban areas. -

Trimethylbenzenes CAS Registry Numbers: 526-73-6 (1,2,3-TMB) 95-63-6 (1,2,4-TMB) 108-67-8 (1,3,5-TMB) 25551-13-7 (Mixed Isomers)
Development Support Document Final, September 4, 2015 Trimethylbenzenes CAS Registry Numbers: 526-73-6 (1,2,3-TMB) 95-63-6 (1,2,4-TMB) 108-67-8 (1,3,5-TMB) 25551-13-7 (Mixed Isomers) Prepared by Joseph T. Haney, Jr., M.S. Angela Curry, M.S. Toxicology Division Office of the Executive Director TEXAS COMMISSION ON ENVIRONMENTAL QUALITY Trimethylbenzenes Page i TABLE OF CONTENTS TABLE OF CONTENTS ............................................................................................................................................ I LIST OF TABLES ......................................................................................................................................................II ACRONYMS AND ABBREVIATIONS ................................................................................................................. III CHAPTER 1 SUMMARY TABLES .......................................................................................................................... 1 CHAPTER 2 MAJOR SOURCES AND USES ......................................................................................................... 4 CHAPTER 3 ACUTE EVALUATION ...................................................................................................................... 4 ACUTE 3.1 HEALTH-BASED ACUTE REV AND ESL ........................................................................................................ 4 3.1.1 Physical/Chemical Properties .................................................................................................................... -

Impact of Molecular Structure on Secondary Organic Aerosol Formation from Aromatic Hydrocarbon
Impact of Molecular Structure on Secondary Organic Aerosol Formation from Aromatic Hydrocarbon Photooxidation under Low NOX Conditions L. Li1,2, P. Tang1,2, S. Nakao1,2,3 and D. R. Cocker III1,2 5 [1] University of California, Riverside, Department of Chemical and Environmental Engineering, Riverside, CA 92507, USA [2] College of Engineering-Center for Environmental Research and Technology (CE- CERT), Riverside, CA 92507, USA [3] Currently at Clarkson University, Department of Chemical and Biomolecular 10 Engineering, Potsdam, NY 13699, USA Correspondence to: D. Cocker III ([email protected]) Abstract The molecular structure of volatile organic compounds (VOC) determines their oxidation pathway, directly impacting secondary organic aerosol (SOA) formation. This study 15 comprehensively investigates the impact of molecular structure on SOA formation from the photooxidation of twelve different eight- to nine-carbon aromatic hydrocarbons under low NOx conditions. The effects of the alkyl substitute number, location, carbon chain length and branching structure on the photooxidation of aromatic hydrocarbons are demonstrated by analyzing SOA yield, chemical composition and physical properties. Aromatic hydrocarbons, 20 categorized into five groups, show a yield order of ortho (o-xylene and o-ethyltoluene)> one substitute (ethylbenzene, propylbenzene and isopropylbenzene) > meta (m-xylene and m- ethyltoluene)> three substitute (trimethylbenzenes) > para (p-xylene and p-ethyltoluene). SOA yields of aromatic hydrocarbon photooxidation do not monotonically decrease when increasing alkyl substitute number. The ortho position promotes SOA formation while the 25 para position suppresses aromatic oxidation and SOA formation. Observed SOA chemical composition and volatility confirm that higher yield is associated with further oxidation. SOA chemical composition also suggests that aromatic oxidation increases with increasing alkyl substitute chain length and branching structure. -
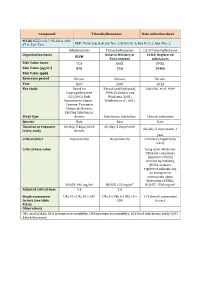
Compound Trimethylbenzenes Data Collection Sheet N°CAS
Compound Trimethylbenzenes Data collection sheet N°CAS 25551-13-7, 95-63-6, 108- 67-8, 526-73-8 CLP: Flam. Liq. 3, Acute Tox. 4, Skin Irrit. 2, Eye Irrit. 2, Asp. Tox. 1, Alkylbenzenes Trimethylbenzenes 1,2,4-Trimethylbenzene Organization Name Ontario Ministry of ECHA: Registered RIVM Environment substances Risk Value Name TCA AAQC DNEL Risk Value (µg/m3) 870 220 29400 Risk Value (ppb) Reference period Chronic Chronic Chronic Year 2007 2007 2013 Key Study Based on Korsak and Rydzynski, Clark DG, et al. 1989 Isopropylbenzene 1996; Gralewicz and EU (2001) Risk Wiaderna, 2001; Assessement Report – Wiaderna et al., 2002 Cumene. European Chemicals Bureau, Existing Substances. Study type chronic Subchronic inhalation Chronic inhalation Species Rats Rats Rats Duration of exposure 6h/day, 5 days/week 6h/day, 5 days/week 6h/day, 5 days/week, 1 in key study chronic year Critical effect Neurotoxicity Neurotoxicity Irritation (respiratory tract) Critical dose value Long term inhalation DNEL for consumers (systemic effects) derived by industry (ECHA-website: registered substances), no transparent information about derivation of DNEL NOAEL 490 mg/m3 NOAEL 123 mg/m³ NOAEC 1800 mg/m3 Adjusted critical dose 5.6 5.6 Single assessment UFH 10 x UFA 10 = 100 UFs 3 x UFH 3 x UFA 10 = 1.7 (Overall assessment factors (see table 100 factor) R.8.6) Other effects UFL used LOAEL; UFH intraspecies variability; UFA interspecies variability; UFS Used subchronic study; UFD data deficiencies Compound TRIMETHYLBENZENES Factsheet Parameter Note Comments Value / descriptor EU-LCI Value and Status EU-LCI value 1 Mass/volume [µg/m³] 450 EU-LCI status 2 Draft / Final Final Year when the EU-LCI value has been EU-LCI year of issue 3 2012 issued General Information 601-025-00-5 CLP-INDEX-Nr. -

Vapor Intrusion Assessments Performed During Site Investigations Petroleum Remediation Program I
www.pca.state.mn.us Vapor intrusion assessments performed during site investigations Petroleum Remediation Program I. Introduction This document describes the methodology for completing a vapor intrusion (VI) assessment at a site in the Minnesota Pollution Control Agency’s (MPCA) Petroleum Remediation Program (PRP). Vapor intrusion is the migration of volatile organic compounds (VOCs) from an underground source through soil or other pathways into buildings. A VI assessment is required when completing a site investigation within PRP. The goal of a VI assessment is to evaluate the VI pathway associated with a petroleum release. The VI pathway has three elements: receptor, vapor source, and subsurface migration route. A receptor is a person potentially affected by VI, generally an occupant of a building. A vapor source is the supply of VOCs, such as contaminated soil or groundwater or light non-aqueous phase liquids (LNAPL). A subsurface migration route is the path that vapors travel through soil or other pathways in the unsaturated zone. The VI assessment uses field-based methods to evaluate the VI pathway, including a receptor survey, soil gas sampling, sub-slab sampling, and indoor air sampling. For additional background information, see Technical Guide For Addressing Petroleum Vapor Intrusion At Leaking Underground Storage Tank Sites (EPA 2015) and Petroleum Vapor Intrusion Fundamentals of Screening, Investigation, and Management (ITRC 2014). II. Step 1 - Soil gas assessment Complete a soil gas assessment while defining the extent and magnitude of soil and groundwater impacts. There are limited situations in which the MPCA may preapprove a different scope of work than what is described here. -

United States Patent Office Patented Jan
3,555,103 United States Patent Office Patented Jan. 12, 1971 2 3,555,103 ter yield with the formation of a smaller number of by PREPARATHON OF CYMENES products, and the fraction of the by-products which may Max Strohmeyer, Ludwigshafen (Rhine), Germany, as be used again in the reaction is higher. In the produc signor to Badische Anilin- & Soda-Fabrik Aktienge tion of p-cymene, far smaller amounts of thr difficultly sellschaft, Ludwigshafen (Rhine), Germany separable o-isomer are obtained. No Drawing. Filed July 16, 1968, Ser. No. 745,093 5 Methyldiisopropylbenzenes, preferably the 1,3,5- and Claims priority, application Germany, July 19, 1967, 1,2,4-methyldiisopropylbenzene, and toluene or mixtures 1,643,629 thereof are used as the starting materials. The aforemen Int. CI. C07c3/58, 5/22 tioned mixtures preferably contain a substance which can U.S. C. 260-672 15 Claims react in the transalkylation stage with toluene to form IO cymenes, for example triisopropylbenzene. The reaction mixture which is obtained at the end of the reaction after ABSTRACT OF THE DISCLOSURE the alkylation stage is advantageously recycled after sepa Process for the preparation of cymenes by reaction of ration of the cymenes, and is used together with fresh methyldiisopropylbenzenes with toluene and propylene in 5 starting materials as the starting mixture. If interest is two or three stages. The products of the new process centered on one specific end product only, for example are valuable starting materials for the production of plas p-cymene, any isomers formed in the reaction may be re tics materials or fibers. -

1,2,4-Trimethylbenzene CAS: 95-63-6 Synonyms: Pseudocumene, Asymmetrical Trimethylbenzene, Psi-Cumene
2009 Risk Assessment Advice for Groundwater Health Risk Assessment Unit, Environmental Health Division 651-201-4899 651-201-5797 TDD Web Publication Date: January 11, 2010 Expiration Date: January 2015 Chemical Name:1,2,4-Trimethylbenzene CAS: 95-63-6 Synonyms: Pseudocumene, Asymmetrical trimethylbenzene, psi-cumene The database for 1,2,4-trimethylbenzene (TMB) consists of 2 short-term oral studies and 5 inhalation studies of varying durations. The currently available data for 1,2,4-trimethylbenzene (TMB) are insufficient to develop chemical specific health-based guidance for groundwater. Health Risk Limits (HRLs) are available for a related trimethylbenzene, 1,3,5-TMB. 1,2,4-TMB and 1,3,5- TMB share similar chemical structures and similar metabolic pathways. A comparison of the available toxicity data for 1,2,4-TMB and 1,3,5-TMB suggests similar toxicity. The use of 1,3,5-TMB-based toxicity values to assess potential health risk from exposure to 1,2,4-TMB has been recommended by US EPA, NCEA (Superfund Program) and Cal EPA, OEHHA. The Minnesota Department of Health (MDH) recommends the use of the HRLs for 1,3,5-TMB to evaluate the potential health risks associated with exposure to 1,2,4-TMB. The HRL values for 1,3,5-TMB are: Acute - Not Derived; Short-term, Subchronic and Chronic - 100 µg/L. For additional information on the derivation of HRL values for 1,3,5-TMB see: http://www.health.state.mn.us/divs/eh/risk/guidance/gw/trimethylbenzene135.pdf . Volatile: Yes (highly volatile) Summary of toxicity testing for health effects identified in the Health Standards Statute: Endocrine Immunotoxicity Development Reproductive Neurotoxicity Tested? No No Yes No Yes Effects? -- Unclear1 Yes2 -- Yes3 (based on 1,3,5- (based on 1,2,4- (based on 1,2,4- trimethylbenzene) trimethylbenzene trimethylbenzene) & 1,3,5- trimethylbenzene) Note: Even if testing for a specific health effect was not conducted for this chemical, information about that effect might be available from studies conducted for other purposes. -

(VOC) Measurements in the Pearl River Delta (PRD) Region, China Ying Liu, Min Shao, Sihua Lu, Chih-Chung Chang, Jia-Lin Wang, Gao Chen
Volatile Organic Compound (VOC) measurements in the Pearl River Delta (PRD) region, China Ying Liu, Min Shao, Sihua Lu, Chih-Chung Chang, Jia-Lin Wang, Gao Chen To cite this version: Ying Liu, Min Shao, Sihua Lu, Chih-Chung Chang, Jia-Lin Wang, et al.. Volatile Organic Compound (VOC) measurements in the Pearl River Delta (PRD) region, China. Atmospheric Chemistry and Physics, European Geosciences Union, 2008, 8 (6), pp.1531-1545. hal-00296488 HAL Id: hal-00296488 https://hal.archives-ouvertes.fr/hal-00296488 Submitted on 13 Mar 2008 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Atmos. Chem. Phys., 8, 1531–1545, 2008 www.atmos-chem-phys.net/8/1531/2008/ Atmospheric © Author(s) 2008. This work is distributed under Chemistry the Creative Commons Attribution 3.0 License. and Physics Volatile Organic Compound (VOC) measurements in the Pearl River Delta (PRD) region, China Ying Liu1, Min Shao1, Sihua Lu1, Chih-chung Chang2, Jia-Lin Wang3, and Gao Chen4 1State Joint Key Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, Beijing, China 2Research Center of Environment Change, Academia Sinica, Nankang, Taipei 115, Taiwan 3Department of Chemistry, National Central University, Chungli 320, Taiwan 4NASA Langley Research Center, Hampton, VA 23681, USA Received: 3 September 2007 – Published in Atmos. -

Analysis of Petroleum Hydrocarbons in Environmental Media
Total Petroleum Hydrocarbon Criteria Working Group Series Volume 1 Analysis of Petroleum Hydrocarbons in Environmental Media Analysis of Petroleum Hydrocarbons in Environmental Media These organizations sponsored or contributed to the completion of this technical document, prepared by the TPH Criteria Working Group: American Petroleum Institute Association for the Environmental Health of Soils Association of American Railroads British Petroleum Chevron Research and Technology Company Exxon Biomedical Sciences, Inc. Retec, Inc. Shell Development Company United States Air Force, Air Force Research Laboratory University of Massachusetts Total Petroleum Hydrocarbon Criteria Working Group Series Volume 1 Analysis of Petroleum Hydrocarbons in Environmental Media March 1998 SPONSORED BY: EDITED BY: Association of American Railroads Wade Weisman BP Oil Company Air Force Research Laboratory, Operational Toxicology Branch United States Air Force, Armstrong Laboratory, Occupational Medicine Division Amherst Scientific Publishers 150 Fearing Street Amherst, Massachusetts 01002 © 1998 by Amherst Scientific Publishers. All rights reserved. ISBN 1-884-940-14-5 The material contained in this document was obtained from independent and highly respected sources. Every attempt has been made to ensure accurate, reliable information, however, the publisher cannot be held responsible for the information or how the information is applied. Opinions expressed in this book are those of the Total Petroleum Hydrocarbon Criteria Working Group and do not reflect those of the publisher. This document was prepared by the Total Petroleum Hydrocarbon Criteria Working Group. Neither the Working Group nor members of the Working Group: a. Makes any warranty or representation, expressed or implied, with respect to the accuracy, com- pleteness, or usefulness of the information contained in this report, or that the use of any appa- ratus, method, or process disclosed in this report may not infringe privately owned rights; or b. -

Environmental Status Overview October 10, 2013
Environmental Status Overview October 10, 2013 Substantial environmental investigation work has already been completed at this site. Tank removals and contaminated material excavations are summarized in the Demolition Summary. This document describes the nature and proposed schedule for ongoing and upcoming environmental work as well the envisioned remedial action strategy. Current Site Contaminants Soil – Predominant chemical compounds detected in site soil at concentrations that exceed WDNR Residual Contaminant Levels (RCLs) include the following: 1. Petroleum compounds ‐ benzene, 1,2‐dichloroethane , ethylbenzene, , naphthalene, toluene, trimethylbenzenes, and xylenes 2. Polynuclear aromatic hydrocarbons (PAHs ) – multiple, see tables 3. Inorganics ‐ arsenic and lead Groundwater – 1. Petroleum – benzene, 1,2‐dichloroethane , ethylbenzene, naphthalene, toluene, trimethylbenzenes, and xylenes 2. PAHs ‐ benzo(a)pyrene, benzo(b)fluoranthene, and chrysene – low concentrations 3. Inorganics – arsenic – relatively low concentrations Chlorinated compounds were generally not detected in site soil or groundwater samples. Upcoming Site Investigation Work Install 2‐3 additional monitoring wells southwest of MW‐10 – late October Complete 2 additional asbestos test pits near sidewalk – late October Remedial Action Strategy and Notes: 4‐Part Remedial Action Strategy supported by WDNR: 1. Excavate/dispose of asbestos contaminated soil (RACM to complete – early November) 2. Vapor Intrusion (VI) Study / Vapor Mitigation Measures (Developer to complete) -

Provisional Peer-Reviewed Toxicity Values for Mixtures of Aliphatic And
EPA/690/R-09/057F l Final 9-30-2009 Provisional Peer-Reviewed Toxicity Values for Complex Mixtures of Aliphatic and Aromatic Hydrocarbons (CASRN Various) Superfund Health Risk Technical Support Center National Center for Environmental Assessment Office of Research and Development U.S. Environmental Protection Agency Cincinnati, OH 45268 Commonly Used Abbreviations BMD Benchmark Dose HI Hazard Index IRIS Integrated Risk Information System IUR inhalation unit risk LOAEL lowest-observed-adverse-effect level LOAELADJ LOAEL adjusted to continuous exposure duration LOAELHEC LOAEL adjusted for dosimetric differences across species to a human NOAEL no-observed-adverse-effect level NOAELADJ NOAEL adjusted to continuous exposure duration NOAELHEC NOAEL adjusted for dosimetric differences across species to a human NOEL no-observed-effect level OSF oral slope factor p-IUR provisional inhalation unit risk p-OSF provisional oral slope factor p-RfC provisional inhalation reference concentration p-RfD provisional oral reference dose p-sRfC provisional subchronic reference concentration p-sRfD provisional subchronic reference dose RfC inhalation reference concentration RfD oral reference dose RPF Relative Potency Factor UF uncertainty factor UFA animal to human uncertainty factor UFC composite uncertainty factor UFD incomplete to complete database uncertainty factor UFH interhuman uncertainty factor UFL LOAEL to NOAEL uncertainty factor UFS subchronic to chronic uncertainty factor i FINAL 9-30-2009 PROVISIONAL PEER-REVIEWED TOXICITY VALUES FOR COMPLEX MIXTURES OF ALIPHATIC AND AROMATIC HYDROCARBONS (CASRN Various) Executive Summary This Provisional Peer-Reviewed Toxicity Value (PPRTV) document supports a fraction-based approach to risk assessment for complex mixtures of aliphatic and aromatic hydrocarbons. The approach takes into account previous efforts, most notably those of the Massachusetts Department of Environmental Protection (MADEP) and the Total Petroleum Hydrocarbon Criteria Working Group (TPHCWG).