PHA 5127 Final Exam Fall 2007
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1: Clinical Pharmacokinetics 1
1: CLINICAL PHARMACOKINETICS 1 General overview: clinical pharmacokinetics, 2 Pharmacokinetics, 4 Drug clearance (CL), 6 Volume of distribution (Vd), 8 The half-life (t½), 10 Oral availability (F), 12 Protein binding (PB), 14 pH and pharmacokinetics, 16 1 Clinical pharmacokinetics General overview General overview: clinical pharmacokinetics 1 The ultimate aim of drug therapy is to achieve effi cacy without toxicity. This involves achieving a plasma concentration (Cp) within the ‘therapeutic window’, i.e. above the min- imal effective concentration (MEC), but below the minimal toxic concentration (MTC). Clinical pharmacokinetics is about all the factors that determine variability in the Cp and its time-course. The various factors are dealt with in subsequent chapters. Ideal therapeutics: effi cacy without toxicity Minimum Toxic Concentration (MTC) Ideal dosing Minimum Effective Concentration (MEC) Drug concentration Time The graph shows a continuous IV infusion at steady state, where the dose-rate is exactly appropriate for the patient’s clearance (CL). Inappropriate dosing Dosing too high in relation to the patient’s CL – toxicity likely Minimum Toxic Concentration (MTC) Minimum Effective Concentration (MEC) Dosing too low in relation to the Drug concentration patient’s CL – drug may be ineffective Time Some reasons for variation in CL Low CL High CL Normal variation Normal variation Renal impairment Increased renal blood fl ow Genetic poor metabolism Genetic hypermetabolism Liver impairment Enzyme induction Enzyme inhibition Old age/neonate 2 General overview Clinical Pharmacokinetics Pharmacokinetic factors determining ideal therapeutics If immediate effect is needed, a loading dose (LD) must be given to achieve a desired 1 concentration. The LD is determined by the volume of distribution (Vd). -

Multidrug-Resistant Gram-Negative Bacteria
Multidrug-Resistant Gram-Negative Bacteria: Trends, Risk Factors, and Treatments A worldwide public health problem, antibiotic resistance leads to treatment-resistant infections associated with prolonged hospitalizations, increased cost, and greater risk for morbidity. Lee S. Engel MD, PhD CASE INTRODUCTION An 80-year-old female nursing home resident with Antibiotics have saved the lives of millions of people a history of dementia, anemia, atrial fibrillation, and have contributed to the major gains in life ex- hypertension, incontinence, and recurrent urinary pectancy over the last century. In US hospitals, 190 tract infections (UTIs), as well as a long-term Foley million doses of antibiotics are administered each catheter, is admitted because she was found to be day.1 Furthermore, more than 133 million courses febrile and less responsive than normal. The patient of antibiotics are prescribed each year for outpatients. has no drug allergies. Upon admission, she has a However, antibiotic use has also resulted in a major temperature of 38.9ºC, heart rate of 92 beats/min, health care challenge—the development and spread and blood pressure of 106/64 mm Hg. The patient of resistant bacteria. Worldwide, antimicrobial resis- opens her eyes to stimuli but does not speak. Aside tance is most evident in diarrheal diseases, respira- from poor dentition and an irregular heart rate, tory tract infections, meningitis, sexually transmitted findings on physical examination, which includes infections, and hospital and health care–acquired in- a pulmonary and abdominal examination, are nor- fections.2 Vancomycin-resistant enterococci, methi- mal. Serum chemistries demonstrate a white blood cillin-resistant Staphylococcus aureus, multidrug-resis- cell (WBC) count of 11,300 cells/mm3. -

No Slide Title
[email protected] In Silico Drug Interaction of Long-acting # 458 Rilpivirine and Cabotegravir With Rifampin Rajith KR Rajoli1, Paul Curley1, David Back1, Charles Flexner2, Andrew Owen1, Marco Siccardi1 1 - Department of Molecular and Clinical Pharmacology, University of Liverpool, Liverpool, UK 2- Johns Hopkins University School of Medicine and Bloomberg School of Public Health, Baltimore, MD, USA Introduction Methods • Physiologically-based pharmacokinetic (PBPK) modelling represents a • Co-administration of anti-TB drugs and many antiretrovirals (ARVs) result mathematical tool to predict DDI magnitude through a detailed • 100 virtual individuals were simulated using Simbiology v.4.3.1, a in important drug-drug interactions (DDIs) understanding of mechanisms underpinning pharmacokinetics product of MATLAB (version 2013b) • Investigation of DDIs between ARVs and anti-TB drugs in individuals can • The objective of this study was to simulate the effect of rifampicin on the • PBPK models were qualified against literature data for oral formulations be complicated by numerous clinical, logistical barriers and also due to pharmacokinetics of cabotegravir and rilpivirine long-acting injectable (LAI) of rifampicin (600 mg OD, at day 6 & 14), cabotegravir and rilpivirine the risk of treatment failure intramuscular (IM) formulations using PBPK modelling (oral, single dose & steady state and IM compared to LATTE-2 studies)1-4 • Loading doses of 800 mg, 900 mg and maintenance doses of 400/800 Results mg, 600/900 mg were used for cabotegravir -

Alcohol Withdrawal
Alcohol withdrawal TERMINOLOGY CLINICAL CLARIFICATION • Alcohol withdrawal may occur after cessation or reduction of heavy and prolonged alcohol use; manifestations are characterized by autonomic hyperactivity and central nervous system excitation 1, 2 • Severe symptom manifestations (eg, seizures, delirium tremens) may develop in up to 5% of patients 3 CLASSIFICATION • Based on severity ○ Minor alcohol withdrawal syndrome 4, 5 – Manifestations occur early, within the first 48 hours after last drink or decrease in consumption 6 □ Manifestations develop about 6 hours after last drink or decrease in consumption and usually peak about 24 to 36 hours; resolution occurs in 2 to 7 days 7 if withdrawal does not progress to major alcohol withdrawal syndrome 4 – Characterized by mild autonomic hyperactivity (eg, tachycardia, hypertension, diaphoresis, hyperreflexia), mild tremor, anxiety, irritability, sleep disturbances (eg, insomnia, vivid dreams), gastrointestinal symptoms (eg, anorexia, nausea, vomiting), headache, and craving alcohol 4 ○ Major alcohol withdrawal syndrome 5, 4 – Progression and worsening of withdrawal manifestations, usually after about 24 hours from the onset of initial manifestations 4 □ Manifestations often peak around 50 hours before gradual resolution or may continue to progress to severe (complicated) withdrawal, particularly without treatment 4 – Characterized by moderate to severe autonomic hyperactivity (eg, tachycardia, hypertension, diaphoresis, hyperreflexia, fever); marked tremor; pronounced anxiety, insomnia, -
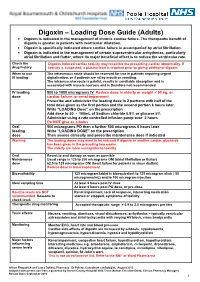
Digoxin – Loading Dose Guide (Adults) Digoxin Is Indicated in the Management of Chronic Cardiac Failure
Digoxin – Loading Dose Guide (Adults) Digoxin is indicated in the management of chronic cardiac failure. The therapeutic benefit of digoxin is greater in patients with ventricular dilatation. Digoxin is specifically indicated where cardiac failure is accompanied by atrial fibrillation. Digoxin is indicated in the management of certain supraventricular arrhythmias, particularly atrial fibrillation and flutter, where its major beneficial effect is to reduce the ventricular rate. Check the Digoxin-induced cardiac toxicity may resemble the presenting cardiac abnormality. If drug history toxicity is suspected, a plasma level is required prior to giving additional digoxin. When to use The intravenous route should be reserved for use in patients requiring urgent IV loading digitalisation, or if patients are nil by mouth or vomiting. The intramuscular route is painful, results in unreliable absorption and is associated with muscle necrosis and is therefore not recommended. IV loading 500 to 1000 micrograms IV Reduce dose in elderly or weight < 50 kg, or dose cardiac failure, or renal impairment Prescribe and administer the loading dose in 2 portions with half of the total dose given as the first portion and the second portion 6 hours later. Write “LOADING Dose” on the prescription Add dose to 50 - 100mL of Sodium chloride 0.9% or glucose 5% Administer using a rate controlled infusion pump over 2 hours Do NOT give as a bolus Oral 500 micrograms PO then a further 500 micrograms 6 hours later loading Write “LOADING DOSE” on the prescription dose Then assess clinically and prescribe maintenance dose if indicated Warning The loading doses may need to be reduced if digoxin or another cardiac glycoside has been given in the preceding two weeks. -
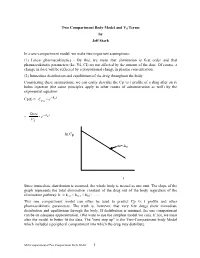
Two Compartment Body Model and Vd Terms by Jeff Stark
Two Compartment Body Model and Vd Terms by Jeff Stark In a one-compartment model, we make two important assumptions: (1) Linear pharmacokinetics - By this, we mean that elimination is first order and that pharmacokinetic parameters (ke, Vd, Cl) are not affected by the amount of the dose. Of course, a change in dose will be reflected by a proportional change in plasma concentration. (2) Immediate distribution and equilibrium of the drug throughout the body. Considering these assumptions, we can easily describe the Cp vs t profile of a drug after an iv bolus injection (the same principles apply to other routes of administration as well) by the exponential equation: − Cp(t) = Ce• kte p0 Dose − = • e kte Vd ln Cp m=-ke t Since immediate distribution is assumed, the whole body is treated as one unit. The slope of the graph represents the total elimination constant of the drug out of the body regardless of the elimination pathway: ke = kren + kmet + kbil + .…. This one compartment model can often be used to predict Cp vs t profile and other pharmacokinetic parameters. The truth is, however, that very few drugs show immediate distribution and equilibrium through the body. If distribution is minimal, the one compartment can be an adequate approximation. (We want to use the simplest model we can). If not, we must alter the model to better fit the data. The "next step up" is the Two-Compartment body Model which includes a peripheral compartment into which the drug may distribute. Multicompartmental/Two Compartment Body Model 1 THE TWO COMPARTMENT MODEL i.v. -

VANCOMYCIN DOSING and MONITORING GUIDELINES (NB Provincial Health Authorities Anti-Infective Stewardship Committee)
Amended: October 2020 VANCOMYCIN DOSING AND MONITORING GUIDELINES (NB Provincial Health Authorities Anti-Infective Stewardship Committee) GENERAL COMMENTS • Vancomycin is a glycopeptide antibiotic with bactericidal activity • It is active against gram-positive bacteria, including methicillin-resistant staphylococcus (MRSA) • Vancomycin is less effective than beta-lactams against Staphylococcus aureus that is susceptible to cloxacillin/methicillin • Vancomycin exhibits time-dependent killing: its effect depends primarily upon the time the concentration exceeds the organism’s Minimum Inhibitory Concentration (MIC) • These guidelines pertain to IV vancomycin only; they do not apply to PO vancomycin, which is not absorbed • Ensure that an adequate mg/kg dose and appropriate interval are ordered initially. Adjust the dose if necessary immediately; do not wait for a confirmatory trough level. • When managing a severe Staphylococcus aureus infection (e.g., bacteremia), an Infectious Diseases consultation is strongly encouraged. VANCOMYCIN IN ADULT PATIENTS ADULT INITIAL DOSE Loading dose: • Consider using a loading dose in patients with: o severe infections where rapid attainment of target level of 10-15 mg/mL is desired o significant renal dysfunction in order to decrease the time required to attain steady state • Recommended dose: 25-30 mg/kg IV o based on actual body weight, for 1 dose, followed by maintenance dose separated by recommended dosing interval o consider capping the loading dose at a maximum of 3g o loading doses DO NOT need to -

Loading Doses in Primary Care
Hull and East Riding Prescribing Committee Preventing fatalities from medication loading doses: Guidance for Primary Care Background A loading dose is an initial large dose of a medicine used to ensure a quick therapeutic response. It is usually given for a short period before therapy continues with a lower maintenance dose. The use of loading doses of medicines can be complex and error prone. Incorrect use of loading doses or subsequent maintenance regimens may lead to severe harm or death. The National Patient Safety Agency (NPSA) issued a Rapid Response Report (NPSA/2010/RRR018) in November 2010, with recommended actions aimed to reduce the number and severity of medication incidents involving incorrect prescribing or administration of loading doses and subsequent maintenance doses. These actions included production of a critical list of medicines, where incorrect loading doses or subsequent maintenance doses are likely to cause harm and ensuring healthcare professionals in primary care are aware of when to challenge abnormal doses of medicines on the agreed critical list. This guidance has been developed as a response to these recommended actions. Critical List Agreed critical list of drugs most likely to cause harm as a result of incorrect prescribing or administration of loading dose or subsequent maintenance dose are: Amiodarone Digoxin Phenytoin Warfarin Best practice in the prescribing, supply and administration of drugs requiring loading dose 1. Where a loading dose is prescribed or recommended, ensure that details of on- going treatment and titration to maintenance dose are clear, and in line with national or local guidelines. 2. Challenge any abnormal prescribing or treatment recommendations (see page 2). -

These Highlights Do Not Include All the Information Needed to Use ACETYLCYSTEINE INJECTION Safely and Effectively
ACETYLCYSTEINE- acetylcysteine injection, solution Fresenius Kabi USA, LLC ---------- HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use ACETYLCYSTEINE INJECTION safely and effectively. See full prescribing information for ACETYLCYSTEINE INJECTION. ACETYLCYSTEINE injection Initial U.S. Approval: 2004. INDICATIONS AND USAGE Acetylcysteine Injection is an antidote for acetaminophen overdose indicated to prevent or lessen hepatic injury after ingestion of a potentially hepatotoxic quantity of acetaminophen (1) DOSAGE AND ADMINISTRATION The total dose of Acetylcysteine Injection is 300 mg/kg given as 3 separate doses and administered over a total of 21 hrs. Please refer to the guidelines below for dose preparation based upon patient weight. Dosing for patients who weigh 5 kg to 20 kg ( 2.1): Loading Dose: 150 mg/kg diluted in 3 mL/kg of diluent* administered over 1 hr Second Dose: 50 mg/kg diluted in 7 mL/kg of diluent* administered over 4 hrs Third Dose: 100 mg/kg diluted in 14 mL/kg of diluent* administered over 16 hrs Dosing for patients who weigh 21 kg to 40 kg ( 2.1): Loading Dose: 150 mg/kg diluted in 100 mL of diluent* administered over 1 hr Second Dose: 50 mg/kg diluted in 250 mL of diluent* administered over 4 hrs Third Dose: 100 mg/kg diluted in 500 mL of diluent* administered over 16 hrs Dosing for patients who weigh 41 kg to 100 kg ( 2.1): Loading Dose: 150 mg/kg diluted in 200 mL of diluent* administered over 1 hr Second Dose: 50 mg/kg diluted in 500mL of diluent* administered over 4 hrs Third Dose: 100 mg/kg diluted in 1,000 mL of diluent* administered over 16 hrs * Acetylcysteine Injection is compatible with the following diluents; 5% Dextrose in Water, 0.45% Sodium Chloride Injection, and Sterile Water for Injection Dosing for patients who weigh more than 100 kg ( 2.1): No specific studies have been conducted to evaluate the use of or necessity of dosing adjustments in patients weighing over 100 kg. -

Simplified Estimation of the Volume of Distribution at Peak Effect for Remifentanil
Simplified Estimation of the Volume of Distribution at Peak Effect for Remifentanil Authors: Elie Sarraf MD, CM; Donald M. Mathews MD Pharmacokinetic mammillary models is described as a central compartment distributing a medications to peripheral compartments and is characterized by volumes of distributions of the compartments as well as their associated clearances. In addition, the volume of distribution at peak effect (Vdpe) [1], is a theoretical volume that relates the pharmacodynamics of the model after a bolus: Loading dose = Cepe * Vdpe Where Cepe is the plasma concentration of the drug at peak effect. For a given patient, the ratio Vdpe to the volume of distribution of the central compartment (V1) is constant and equal to the ratio between the initial plasma concentration (C0) after a bolus to Cepe. This ratio can be determined through simulation by directly calculating C0/Cepe or through non-trivial arithmetic computations. We propose an alternative approach to determining the ratio through parameter estimation which we apply to Minto’s remifentanil model [2]. This approach results in a simple method to calculate the ratio and thus Vdpe. Methods: Using Matlab (R2016b), the Cepe along with the C0 was determined for simulated patient between the ages of 20 and 80 in increments of 5 years, weights of 40 and 120 kg in increments of 5, heights of 120 and 220 cm in increments of 10 cm, as well as both genders. Patients with lean body mass (LBM) below 20 kg were excluded from further analysis. Age, weight, height, gender, LBM, volumes of distribution (V1, V2 and V3), clearances (Cl1, Cl2, Cl3), ke0 and the ratio C0/Cepe were tabulated. -

Lanoxin, Tablet
NEW ZEALAND DATA SHEET 1. LANOXIN Digoxin tablets 62.5mcg (0.0625mg) and 250mcg (0.250mg); Paediatric Elixir 50mcg/mL 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Tablets: Each tablet contains 62.5mcg (0.0625mg), 250mcg (0.250mg) Digoxin BP Paediatric Elixir: 50mcg Digoxin BP in each 1mL For full list of excipients, see section 6.1 3. PHARMACEUTICAL FORM Tablets PG 62.5mcg Blue, round, biconvex tablets debossed “DO6” and each containing 62.5mcg (0.0625mg) Digoxin BP. Tablets 250mcg White, round, biconvex tablets, bisected and debossed ‘DO25” and each containing 250mcg (0.250mg) Digoxin BP. Paediatric Elixir 50mcg/mL A clear, yellow, lime flavoured solution containing 50mcg (0.050mg) Digoxin BP in each 1mL of sweetened, aqueous-alcoholic vehicle. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Cardiac Failure LANOXIN is indicated in the management of chronic cardiac failure where the dominant problem is systolic dysfunction. Its therapeutic benefit is greatest in those patients with ventricular dilatation. LANOXIN is specifically indicated where cardiac failure is accompanied by atrial fibrillation. Supraventricular Arrhythmias LANOXIN is indicated in the management of certain supraventricular arrhythmias, particularly atrial flutter and fibrillation, where a major beneficial effect is reduction of the ventricular rate. 4.2 Dose and method of administration The dose of LANOXIN for each patient has to be tailored individually according to age, lean body weight and renal function. Suggested doses are intended only as an initial guide. LANOXIN PG Oral Solution, 50mcg in 1mL, is supplied with a graduated pipette and this should be used for measurement of all doses. Adults and children over 10 years Rapid Oral Loading 1 If medically appropriate, rapid digitalisation may be achieved in a number of ways, such as the following: 750 to 1500mcg (0.75 to 1.5mg) as a single dose. -

Preventing Phenytoin Intoxication: Safer Use of a Familiar Anticonvulsant
Applied Evidence N EW R ESEARCH F INDINGS T HAT A RE C HANGING C LINICAL P RACTICE Preventing phenytoin intoxication: Safer use of a familiar anticonvulsant Thomas H. Glick, MD, Tom P.Workman, MD, and Slava V. Gaufberg, MD Cambridge Health Alliance, Cambridge, Mass; Harvard Medical School, Boston, Mass Practice recommendations ■ Safe practice for initiating or adjusting a maintenance dosage should include patient ■ To load phenytoin initially or to add a education and close follow-up (C). supplemental load, use this formula: for each µg/mL desired increase in the phenytoin serum level, increase the espite the introduction of new anticonvul- loading dose by 0.75 mg/kg (C). sant drugs, phenytoin is still a first-line medication for common types of epileptic ■ Measure the peak serum level shortly D seizures, particularly those caused by focal brain after loading—30 to 60 minutes or more lesions.1–4 Available in parenteral and oral form, after giving intravenous phenytoin, phenytoin (or its pro-drug, fosphenytoin) is widely 2 hours or more after intravenous used. An estimated 873,000 prescriptions for fosphenytoin, 4 hours or more after phenytoin were issued during office visits in 2001.5 intramuscular fosphenytoin, and 16–24 Phenytoin carries a special risk of dose-related hours after accelerated oral loading (C). toxicity, due to its saturation (zero-order) phar- ■ The daily maintenance dose (mg/kg/d) macokinetics: serum levels often rise much ordinarily needed to achieve a specified more than would ordinarily be expected after serum level or maintain it after loading is initiating or increasing a maintenance dose.