Loading Dose of Drug A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1: Clinical Pharmacokinetics 1
1: CLINICAL PHARMACOKINETICS 1 General overview: clinical pharmacokinetics, 2 Pharmacokinetics, 4 Drug clearance (CL), 6 Volume of distribution (Vd), 8 The half-life (t½), 10 Oral availability (F), 12 Protein binding (PB), 14 pH and pharmacokinetics, 16 1 Clinical pharmacokinetics General overview General overview: clinical pharmacokinetics 1 The ultimate aim of drug therapy is to achieve effi cacy without toxicity. This involves achieving a plasma concentration (Cp) within the ‘therapeutic window’, i.e. above the min- imal effective concentration (MEC), but below the minimal toxic concentration (MTC). Clinical pharmacokinetics is about all the factors that determine variability in the Cp and its time-course. The various factors are dealt with in subsequent chapters. Ideal therapeutics: effi cacy without toxicity Minimum Toxic Concentration (MTC) Ideal dosing Minimum Effective Concentration (MEC) Drug concentration Time The graph shows a continuous IV infusion at steady state, where the dose-rate is exactly appropriate for the patient’s clearance (CL). Inappropriate dosing Dosing too high in relation to the patient’s CL – toxicity likely Minimum Toxic Concentration (MTC) Minimum Effective Concentration (MEC) Dosing too low in relation to the Drug concentration patient’s CL – drug may be ineffective Time Some reasons for variation in CL Low CL High CL Normal variation Normal variation Renal impairment Increased renal blood fl ow Genetic poor metabolism Genetic hypermetabolism Liver impairment Enzyme induction Enzyme inhibition Old age/neonate 2 General overview Clinical Pharmacokinetics Pharmacokinetic factors determining ideal therapeutics If immediate effect is needed, a loading dose (LD) must be given to achieve a desired 1 concentration. The LD is determined by the volume of distribution (Vd). -
Prediction of Clinical Transporter‐Mediated Drug–Drug Interactions
Citation: CPT Pharmacometrics Syst. Pharmacol. (2020) 9, 211–221; doi:10.1002/psp4.12505 ARTICLE Prediction of Clinical Transporter-Mediated Drug–Drug Interactions via Comeasurement of Pitavastatin and Eltrombopag in Human Hepatocyte Models Simon J. Carter1, Bhavik Chouhan2, Pradeep Sharma3 and Michael J. Chappell1,* A structurally identifiable micro-rate constant mechanistic model was used to describe the interaction between pitavastatin and eltrombopag, with improved goodness-of-fit values through comeasurement of pitavastatin and eltrombopag. Transporter association and dissociation rate constants and passive rates out of the cell were similar between pitavastatin and eltrom- bopag. Translocation into the cell through transporter-mediated uptake was six times greater for pitavastatin, leading to pronounced inhibition of pitavastatin uptake by eltrombopag. The passive rate into the cell was 91 times smaller for pitavas- tatin compared with eltrombopag. A semimechanistic physiologically-based pharmacokinetic (PBPK) model was developed to evaluate the potential for clinical drug–drug interactions (DDIs). The PBPK model predicted a twofold increase in the pita- vastatin peak blood concentration and area under the concentration-time curve in the presence of eltrombopag in simulated healthy volunteers. The use of structural identifiability supporting experimental design combined with robust micro-rate con- stant parameter estimates and a semimechanistic PBPK model gave more informed predictions of transporter-mediated DDIs. Study Highlights WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? model fits through micro-rate constants compared with ✔ Currently, most in vitro models are not guided by macro-rate constants in human hepatocytes, with addi- structural identifiability analysis, relying on substrate-only tional information provided regarding transporter binding. -

Multidrug-Resistant Gram-Negative Bacteria
Multidrug-Resistant Gram-Negative Bacteria: Trends, Risk Factors, and Treatments A worldwide public health problem, antibiotic resistance leads to treatment-resistant infections associated with prolonged hospitalizations, increased cost, and greater risk for morbidity. Lee S. Engel MD, PhD CASE INTRODUCTION An 80-year-old female nursing home resident with Antibiotics have saved the lives of millions of people a history of dementia, anemia, atrial fibrillation, and have contributed to the major gains in life ex- hypertension, incontinence, and recurrent urinary pectancy over the last century. In US hospitals, 190 tract infections (UTIs), as well as a long-term Foley million doses of antibiotics are administered each catheter, is admitted because she was found to be day.1 Furthermore, more than 133 million courses febrile and less responsive than normal. The patient of antibiotics are prescribed each year for outpatients. has no drug allergies. Upon admission, she has a However, antibiotic use has also resulted in a major temperature of 38.9ºC, heart rate of 92 beats/min, health care challenge—the development and spread and blood pressure of 106/64 mm Hg. The patient of resistant bacteria. Worldwide, antimicrobial resis- opens her eyes to stimuli but does not speak. Aside tance is most evident in diarrheal diseases, respira- from poor dentition and an irregular heart rate, tory tract infections, meningitis, sexually transmitted findings on physical examination, which includes infections, and hospital and health care–acquired in- a pulmonary and abdominal examination, are nor- fections.2 Vancomycin-resistant enterococci, methi- mal. Serum chemistries demonstrate a white blood cillin-resistant Staphylococcus aureus, multidrug-resis- cell (WBC) count of 11,300 cells/mm3. -

624.Full.Pdf
1521-009X/44/5/624–633$25.00 http://dx.doi.org/10.1124/dmd.115.068932 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 44:624–633, May 2016 Copyright ª 2016 by The American Society for Pharmacology and Experimental Therapeutics Development of a Rat Plasma and Brain Extracellular Fluid Pharmacokinetic Model for Bupropion and Hydroxybupropion Based on Microdialysis Sampling, and Application to Predict Human Brain Concentrations Thomas I.F.H. Cremers, Gunnar Flik, Joost H.A. Folgering, Hans Rollema, and Robert E. Stratford, Jr. Brains On-Line BV, Groningen, The Netherlands (T.I.F.H.C., G.F. J.H.A.F.); Rollema Biomedical Consulting, Mystic, Connecticut (H.R.); and Division of Pharmaceutical Sciences, Mylan School of Pharmacy, Duquesne University, Pittsburgh, Pennsylvania (R.E.S.) Received December 11, 2015; accepted February 24, 2016 Downloaded from ABSTRACT Administration of bupropion [(6)-2-(tert-butylamino)-1-(3-chlorophenyl) concentration ratio at early time points was observed with propan-1-one] and its preformed active metabolite, hydroxybupropion bupropion; this was modeled as a time-dependent uptake clear- [(6)-1-(3-chlorophenyl)-2-[(1-hydroxy-2-methyl-2-propanyl)amino]- ance of the drug across the blood–brain barrier. Translation of the 1-propanone], to rats with measurement of unbound concentrations model was used to predict bupropion and hydroxybupropion dmd.aspetjournals.org by quantitative microdialysis sampling of plasma and brain extra- exposure in human brain extracellular fluid after twice-daily cellular fluid was used to develop a compartmental pharmacoki- administration of 150 mg bupropion. Predicted concentrations netics model to describe the blood–brain barrier transport of both indicate that preferential inhibition of the dopamine and norepi- substances. -

A Primer on Pharmacology
A primer on pharmacology Universidade do Algarve Faro 2017 by Ferdi Engels, Ph.D. 1 2 1 3 Utrecht university campus ‘de Uithof’ Dept. of Pharmaceutical Sciences Division of Pharmacology 4 2 Bachelor and master education Ferdi Engels, PhD ‐ Associate professor of pharmacology ‐ Director of Undergraduate School of Science PhD training Research expertise 5 for today 1. Understand the main concepts of pharmacokinetics Main concept 2. Be able to apply this new knowledge 6 3 Pharmacology is about drugs………. Drugs = chemicals that alter physiological processes in the body for treatment, prevention, or cure of diseases input output (administration of the drug) (biological response) ‐ dose ‐ no effect ‐ frequency of administration ‐ beneficial effects ‐ route of administration ‐ adverse / toxic effects onset, intensity, and duration of therapeutic effects 7 Pharmacology is about drugs………. Drugs = chemicals that alter physiological processes in the body for treatment, prevention, or cure of diseases What does the body do to the drug? pharmacokinetics What does the drug do to the body? pharmacodynamics 8 4 Thursday, June 15 Lecture on topic 1 Workshop on topic 1 Friday, June 16 Lecture on topic 2 Workshop on topic 2 Course Topic 1 Course Topic 2 9 Rosenbaum ‐ Basic pharmacokinetics and pharmacodynamics: an integrated textbook and computer simulations, 1st ed. (2011) input output (administration of the drug) (biological response) ‐ dose ‐ no effect ‐ frequency of administration ‐ beneficial effects ‐ route of administration ‐ adverse / toxic effects -

Mechanism-Based Inactivation of Human Cytochrome P450 Enzymes and the Prediction of Drug-Drug Interactions□S
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2006/11/08/dmd.106.012633.DC1.html 0090-9556/07/3502-246–255$20.00 DRUG METABOLISM AND DISPOSITION Vol. 35, No. 2 Copyright © 2007 by The American Society for Pharmacology and Experimental Therapeutics 12633/3169778 DMD 35:246–255, 2007 Printed in U.S.A. Mechanism-Based Inactivation of Human Cytochrome P450 Enzymes and the Prediction of Drug-Drug Interactions□S R. Scott Obach, Robert L. Walsky, and Karthik Venkatakrishnan Pharmacokinetics, Dynamics, and Drug Metabolism (R.S.O., R.L.W.) and Clinical Pharmacology (K.V.), Pfizer, Inc., Groton, Connecticut Received August 30, 2006; accepted November 2, 2006 ABSTRACT: The ability to use vitro inactivation kinetic parameters in scaling to proaches to identifying mechanism-based inactivators were devel- in vivo drug-drug interactions (DDIs) for mechanism-based inacti- oped. Testing potential inactivators at a single concentration (IC25) vators of human cytochrome P450 (P450) enzymes was examined in a 30-min preincubation with human liver microsomes in the Downloaded from using eight human P450-selective marker activities in pooled hu- absence and presence of NADPH followed by assessment of P450 man liver microsomes. These data were combined with other pa- marker activities readily identified those compounds known to be rameters (systemic Cmax, estimated hepatic inlet Cmax, fraction mechanism-based inactivators and represents an approach that unbound, in vivo P450 enzyme degradation rate constants -

1. Disposition and Pharmacokinetics
1. DISPOSITION AND PHARMACOKINETICS The disposition and pharmacokinetics of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and related compounds have been investigated in several species and under various exposure conditions. Several reviews on this subject focus on TCDD and related halogenated aromatic hydrocarbons (Neal et al., 1982; Gasiewicz et al., 1983a; Olson et al., 1983; Birnbaum, 1985; van den Berg et al., 1994). The relative biological and toxicological potency of TCDD and related compounds depends not only on the affinity of these compounds for the aryl hydrocarbon receptor (AhR), but on the species-, strain-, and congener-specific pharmacokinetics of these compounds (Neal et al., 1982; Gasiewicz et al., 1983a; Olson et al., 1983; Birnbaum, 1985; van den Berg et al., 1994, DeVito and Birnbaum, 1995). 2,3,7,8-TCDD and other similar compounds discussed here are rapidly absorbed into the body and slowly eliminated, making body burden (bioaccumulation) a reliable indicator of time- integrated exposure and absorbed dose. Because of the slow elimination kinetics, it will be shown in this section that lipid or blood concentrations, which are often measured, are in dynamic equilibrium with other tissue compartments in the body, making the overall body burden and tissue disposition relatively easy to estimate. Finally, it will be shown that body burdens can be correlated with adverse health effects (Hardell et al., 1995; Leonards et al., 1995), further leading to the choice of body burden as the optimal indicator of absorbed dose and potential effects. 1.1. ABSORPTION/BIOAVAILABILITY FOLLOWING EXPOSURE Gastrointestinal, dermal, and transpulmonary absorptions represent potential routes for human exposure to this class of persistent environmental contaminants. -

The Impact of Assay Acceptance Criteria on Derived Data – Pharmacokinetic Assessment Through Simulation
The Impact of Assay Acceptance Criteria on Derived Data – Pharmacokinetic Assessment Through Simulation Oriol Peris – Charles River Laboratories EBF 2021 EVERY STEP OF THE WAY EVERY STEP OF THE WAY CONFIDENTIAL Objective • Samples analysed using chromatographic assays have an acceptance criteria for QCs of ±15%. In contrast, ligand binding assays have an acceptance criteria for QCs of ±20% • This could potentially lead to a greater variability introduced in samples analysed using ligand binding assays, compared to chromatographic assays. • The aim of this presentation is to assess, from a PK perspective, the impact that the variability associated to each of these methodologies may have in the assessment of the data. To do this: • Plausible data with and without variability needs to be generated • The generated data needs to be assessed using an objective method Methodology: Data generation • Concentration vs time profiles were simulated using open source R software and its library mrgsolve, under different scenarios • Low between subject variability (BSV) and 15% acceptance criterion for QCs • High BSV and 15% acceptance criterion for QCs • Low BSV and 20% acceptance criterion for QCs • High BSV and 20% acceptance criterion for QCs • To do this, it was assumed that all chromatographic and ligand binding assays may introduce up to ± 15 or ± 20% deviation from nominal concentrations • The data was simulated using a population 1 compartment extravascular PK model with parameters CL, Vd and Ka #$%$& • 1 compartment extravascular model � = ' �*&+$- − �*&'$- () &'*&+ Methodology: Data generation • PK compartmental models: • Set of exponential equations that are useful to model a concentration vs time profile • Define the body as divided in separate compartments where a compartment can be the body, blood circulatory system or a set of tissues or organs sharing similar affinity to a drug • PK compartmental models are useful as they are mathematically plausible and their parameters (i.e. -

No Slide Title
[email protected] In Silico Drug Interaction of Long-acting # 458 Rilpivirine and Cabotegravir With Rifampin Rajith KR Rajoli1, Paul Curley1, David Back1, Charles Flexner2, Andrew Owen1, Marco Siccardi1 1 - Department of Molecular and Clinical Pharmacology, University of Liverpool, Liverpool, UK 2- Johns Hopkins University School of Medicine and Bloomberg School of Public Health, Baltimore, MD, USA Introduction Methods • Physiologically-based pharmacokinetic (PBPK) modelling represents a • Co-administration of anti-TB drugs and many antiretrovirals (ARVs) result mathematical tool to predict DDI magnitude through a detailed • 100 virtual individuals were simulated using Simbiology v.4.3.1, a in important drug-drug interactions (DDIs) understanding of mechanisms underpinning pharmacokinetics product of MATLAB (version 2013b) • Investigation of DDIs between ARVs and anti-TB drugs in individuals can • The objective of this study was to simulate the effect of rifampicin on the • PBPK models were qualified against literature data for oral formulations be complicated by numerous clinical, logistical barriers and also due to pharmacokinetics of cabotegravir and rilpivirine long-acting injectable (LAI) of rifampicin (600 mg OD, at day 6 & 14), cabotegravir and rilpivirine the risk of treatment failure intramuscular (IM) formulations using PBPK modelling (oral, single dose & steady state and IM compared to LATTE-2 studies)1-4 • Loading doses of 800 mg, 900 mg and maintenance doses of 400/800 Results mg, 600/900 mg were used for cabotegravir -

Alcohol Withdrawal
Alcohol withdrawal TERMINOLOGY CLINICAL CLARIFICATION • Alcohol withdrawal may occur after cessation or reduction of heavy and prolonged alcohol use; manifestations are characterized by autonomic hyperactivity and central nervous system excitation 1, 2 • Severe symptom manifestations (eg, seizures, delirium tremens) may develop in up to 5% of patients 3 CLASSIFICATION • Based on severity ○ Minor alcohol withdrawal syndrome 4, 5 – Manifestations occur early, within the first 48 hours after last drink or decrease in consumption 6 □ Manifestations develop about 6 hours after last drink or decrease in consumption and usually peak about 24 to 36 hours; resolution occurs in 2 to 7 days 7 if withdrawal does not progress to major alcohol withdrawal syndrome 4 – Characterized by mild autonomic hyperactivity (eg, tachycardia, hypertension, diaphoresis, hyperreflexia), mild tremor, anxiety, irritability, sleep disturbances (eg, insomnia, vivid dreams), gastrointestinal symptoms (eg, anorexia, nausea, vomiting), headache, and craving alcohol 4 ○ Major alcohol withdrawal syndrome 5, 4 – Progression and worsening of withdrawal manifestations, usually after about 24 hours from the onset of initial manifestations 4 □ Manifestations often peak around 50 hours before gradual resolution or may continue to progress to severe (complicated) withdrawal, particularly without treatment 4 – Characterized by moderate to severe autonomic hyperactivity (eg, tachycardia, hypertension, diaphoresis, hyperreflexia, fever); marked tremor; pronounced anxiety, insomnia, -
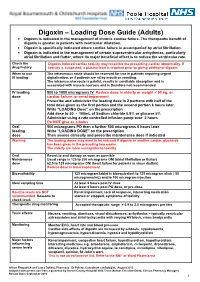
Digoxin – Loading Dose Guide (Adults) Digoxin Is Indicated in the Management of Chronic Cardiac Failure
Digoxin – Loading Dose Guide (Adults) Digoxin is indicated in the management of chronic cardiac failure. The therapeutic benefit of digoxin is greater in patients with ventricular dilatation. Digoxin is specifically indicated where cardiac failure is accompanied by atrial fibrillation. Digoxin is indicated in the management of certain supraventricular arrhythmias, particularly atrial fibrillation and flutter, where its major beneficial effect is to reduce the ventricular rate. Check the Digoxin-induced cardiac toxicity may resemble the presenting cardiac abnormality. If drug history toxicity is suspected, a plasma level is required prior to giving additional digoxin. When to use The intravenous route should be reserved for use in patients requiring urgent IV loading digitalisation, or if patients are nil by mouth or vomiting. The intramuscular route is painful, results in unreliable absorption and is associated with muscle necrosis and is therefore not recommended. IV loading 500 to 1000 micrograms IV Reduce dose in elderly or weight < 50 kg, or dose cardiac failure, or renal impairment Prescribe and administer the loading dose in 2 portions with half of the total dose given as the first portion and the second portion 6 hours later. Write “LOADING Dose” on the prescription Add dose to 50 - 100mL of Sodium chloride 0.9% or glucose 5% Administer using a rate controlled infusion pump over 2 hours Do NOT give as a bolus Oral 500 micrograms PO then a further 500 micrograms 6 hours later loading Write “LOADING DOSE” on the prescription dose Then assess clinically and prescribe maintenance dose if indicated Warning The loading doses may need to be reduced if digoxin or another cardiac glycoside has been given in the preceding two weeks. -
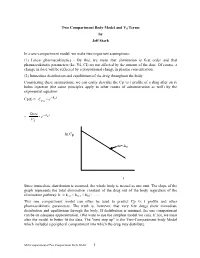
Two Compartment Body Model and Vd Terms by Jeff Stark
Two Compartment Body Model and Vd Terms by Jeff Stark In a one-compartment model, we make two important assumptions: (1) Linear pharmacokinetics - By this, we mean that elimination is first order and that pharmacokinetic parameters (ke, Vd, Cl) are not affected by the amount of the dose. Of course, a change in dose will be reflected by a proportional change in plasma concentration. (2) Immediate distribution and equilibrium of the drug throughout the body. Considering these assumptions, we can easily describe the Cp vs t profile of a drug after an iv bolus injection (the same principles apply to other routes of administration as well) by the exponential equation: − Cp(t) = Ce• kte p0 Dose − = • e kte Vd ln Cp m=-ke t Since immediate distribution is assumed, the whole body is treated as one unit. The slope of the graph represents the total elimination constant of the drug out of the body regardless of the elimination pathway: ke = kren + kmet + kbil + .…. This one compartment model can often be used to predict Cp vs t profile and other pharmacokinetic parameters. The truth is, however, that very few drugs show immediate distribution and equilibrium through the body. If distribution is minimal, the one compartment can be an adequate approximation. (We want to use the simplest model we can). If not, we must alter the model to better fit the data. The "next step up" is the Two-Compartment body Model which includes a peripheral compartment into which the drug may distribute. Multicompartmental/Two Compartment Body Model 1 THE TWO COMPARTMENT MODEL i.v.