Synthetic Investigation of Natural Products Causing Dopaminergic Neurodegeneration
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![C9-14 Aliphatic [2-25% Aromatic] Hydrocarbon Solvents Category SIAP](https://docslib.b-cdn.net/cover/2852/c9-14-aliphatic-2-25-aromatic-hydrocarbon-solvents-category-siap-12852.webp)
C9-14 Aliphatic [2-25% Aromatic] Hydrocarbon Solvents Category SIAP
CoCAM 2, 17-19 April 2012 BIAC/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical C -C Aliphatic [2-25% aromatic] Hydrocarbon Solvents Category Category 9 14 Substance Name CAS Number Stoddard solvent 8052-41-3 Chemical Names Kerosine, petroleum, hydrodesulfurized 64742-81-0 and CAS Naphtha, petroleum, hydrodesulfurized heavy 64742-82-1 Registry Solvent naphtha, petroleum, medium aliphatic 64742-88-7 Numbers Note: Substances in this category are also commonly known as mineral spirits, white spirits, or Stoddard solvent. CAS Number Chemical Description † 8052-41-3 Includes C8 to C14 branched, linear, and cyclic paraffins and aromatics (6 to 18%), <50ppmV benzene † 64742-81-0 Includes C9 to C14 branched, linear, and cyclic paraffins and aromatics (10 to Structural 25%), <100 ppmV benzene Formula † and CAS 64742-82-1 Includes C8 to C13 branched, linear, and cyclic paraffins and aromatics (15 to 25%), <100 ppmV benzene Registry † Numbers 64742-88-7 Includes C8 to C13 branched, linear, and cyclic paraffins and aromatics (14 to 20%), <50 ppmV benzene Individual category member substances are comprised of aliphatic hydrocarbon molecules whose carbon numbers range between C9 and C14; approximately 80% of the aliphatic constituents for a given substance fall within the C9-C14 carbon range and <100 ppmV benzene. In some instances, the carbon range of a test substance is more precisely defined in the test protocol. In these instances, the specific carbon range (e.g. C8-C10, C9-C10, etc.) will be specified in the SIAP. * It should be noted that other substances defined by the same CAS RNs may have boiling ranges outside the range of 143-254° C and that these substances are not covered by the category. -

Long-Term Strategy for Russian Olive and Saltcedar Management
LONG-TERM STRATEGY FOR RUSSIAN OLIVE AND SALTCEDAR MANAGEMENT YELLOWSTONE RIVER CONSERVATION DISTRICT COUNCIL Prepared by Thomas L. Pick, Bozeman, Montana May 1, 2013 Cover photo credit: Tom Pick. Left side: Plains cottonwood seedlings on bar following 2011 runoff. Top: Saltcedar and Russian olive infest the shoreline of the Yellowstone River between Hysham and Forsyth, Montana. Bottom: A healthy narrowleaf cottonwood (Populus angustifolia James) stand adjacent to the channel provides benefits for wildlife and livestock in addition to bank stability and storing floodwater for later release. Table of Contents page number Agreement ………………………………………………………………………………………………………………………………………….1 Executive Summary …………………………………………………………………………………………………………………………….2 Long-Term Strategy for Russian Olive and Saltcedar Management 1.0 Introduction ………………………………………………………………………………………………………………………4 1.1 Biology and Ecology of Russian Olive and Saltcedar ……………………………………………………5 1.12 Russian Olive 1.13 Saltcedar 1.2 Distribution and Spread……………………………………………………………………………………………..6 1.3 Summary of Impacts ………………………………………………………………………………………………….6 1.4 Legal Framework ……………………………………………………………………………………………………….7 2.0 Strategic Management Objective and Goals ………………………………………………………………..…..8 2.1 Goal 1: Prevent New Infestations (Control Spread) …………………………………….……….……8 2.2 Goal 2: Eradicate All Infestations Within the River Corridor ………………………………………9 2.3 Goal 3: Manage Populations Outside of the River Corridor ……………………………………….9 3.0 Treatment Strategies and Priorities …………………………………….……………………………………………9 -

BULLETIN for the HISTORY of CHEMISTRY Division of the History of Chemistry of the American Chemical Society
BULLETIN FOR THE HISTORY OF CHEMISTRY Division of the History of Chemistry of the American Chemical Society VOLUME 29, Number 1 2004 BULLETIN FOR THE HISTORY OF CHEMISTRY VOLUME 29, CONTENTS NUMBER 1 THE 2003 EDELSTEIN AWARD ADDRESS* MAKING CHEMISTRY POPULAR David Knight, University of Durham, England 1 THE DISCOVERY OF LECITHIN, THE FIRST PHOSPHOLIPID Theodore L. Sourkes, McGill University 9 GABRIEL LIPPMANN AND THE CAPILLARY ELECTROMETER John T. Stock, University of Connecticut 16 KHEMYE: CHEMICAL LITERATURE IN YIDDISH Stephen M. Cohen 21 AN EARLY HISTORY OF CHEMISTRY AT TEXAS TECH UNIVERSITY, 1925-1970* Henry J. Shine, Texas Tech University 30 NOYES LABORATORY, AN ACS NATIONAL CHEMICAL LANDMARK: 100 YEARS OF CHEMISTRY AT THE UNIVERSITY OF ILLINOIS Sharon Bertsch McGrayne 45 BOOK REVIEWS 52 The Cover…….See page 24. Bull. Hist. Chem., VOLUME 29, Number 1 (2004) 1 THE 2003 EDELSTEIN AWARD ADDRESS* MAKING CHEMISTRY POPULAR David Knight, University of Durham, England “Chemistry is wonderful,” wrote evenings, and a bright dawn Linus Pauling (1), “I feel sorry for gleamed over a chemically-based people who don’t know anything society. Intellectually, the science about chemistry. They are miss- did not demand the mathematics re- ing an important source of happi- quired for serious pursuit of the sub- ness.” That is not how the science lime science of astronomy. Chem- has universally been seen in our ists like Joseph Priestley thought it time. We would not expect to see the ideal Baconian science in which lecture-rooms crowded out, chem- everyone might join, for its theoreti- ists as stars to be invited to fash- cal structure was still unformed. -

Personal View – the Evolution of Neurochemistry
Neuroforum 2019; 25(4): 259–264 Review Article Ferdinand Hucho* Personal View – The Evolution of Neurochemistry Two questions – one answer https://doi.org/10.1515/nf-2019-0023 century up to our times. Reductionism was hoped to solve two of the most fundamental riddles which were central to Abstract: This esssay is a personal account of the evolution human thinking since antiquity: The world, believed to be of Neurochemistry in the past century. It describes in par- composed of ‚mind and matter‘, poses the question: What allel the authors way from chemistry to biochemistry and is life? The neurochemist goes one step further and asks: finally to Neurochemistry and the progress of a most ex- what is mind (conciousness, cognition, free will)? The citing chapter of the Life Sciences. It covers the successful physiologist Emil du Bois-Reymond (1818–1896) included time period of reductionist research (by no means compre- these questions in his ‚seven riddles‘ (Finkelstein 2013) hensively), which lay the ground for the recent and future and summarized his answer in 1880 in his famous “igno- systems approach. This development promises answers to ramus et ignorabimus” (“we don‘t know and we never will fundamental questions of our existence as human beings. know”). Never say ‘never’, because this could be the end Keywords: Chemistry; Biochemistry; Life Science; Neuro- of human curiosity and research, preventing discoveries chemistry including new methods of investigation. In the 20th century the question What is life was most vividly posed by physicists like the Nobel laureates Zusammenfassung: Dieser Essay ist ein persönlicher Erwin Schrödinger (Schrödinger 1944; Fischer ed., 1987) Bericht über die Entwicklung der Neurochemie im ver- and Max Delbrück (Delbrück 1986). -

Essays in Neurochemistry and Neuropharmacology Chemical
Volume 89, number 2 FEBS LETTERS May 1978 Essays in Neurochemistry and Neuropharmacology Volume 2 Edited by M. B. H. Youdim, W. Lovenberg, D. F. Sharman and J. R. Lagnado John Wiley and Sons; Chichester, New York, Brisbane, Toronto, 1977 xiv + 174 pages, £8.75 An essay on essays prefaces this book, praising the The Biochemical Society's 'Essays in Biochemistry' literary essay and advocating a comparable form of which antedate the present series by I 1 years are also scientific writing as a leavening or antidote to the despite their name broad, didactically-oriented reviews stereotype of papers proceeding by Methods-Results- in which many authors use figures and tables as a Discussion. The terse wisdom and precise word-choise means of avoiding cumbersome passages of text; a of Bacon, or the charm of Lamb, would indeed be necessary distinction from the literary essay and much welcome in the neurosciences, but this book's to be encouraged. So also are author and subject contents exemplify a different and quite worthy indexes which are lacking in the present book. It also stereotype, the review. Are literary essays ever multi- has no running titles or author's names at the head of author, as are half of the present contributions? Two its pages; and as the list of abbrevations and the of the most effective essays here are indeed single- literature references come at the beginning and end of authored: the first few pages of K. G. Walton's each individual essay, lack of this guidance to where account of cyclic necleotides and postynaptic events the beginning and end are located is to be regretted. -

Acupuncture and Traditional Chinese Medicine in the Treatment of Parkinson’S Disease
Schulz Capstone Acupuncture and Traditional Chinese Medicine In the Treatment of Parkinson’s Disease By Mary M. Schulz Presented in partial fulfillment for the Doctor of Acupuncture and Oriental Medicine Degree Capstone Advisors: Eric Tamrazian, M.D., Lawrence J. Ryan, Ph.D. Yo San University April 2014 Schulz Capstone Approvals Signatures Page This Capstone Project has been reviewed and approved by: _________________________________________ __5/4/2015__________ Eric Tamrazian, M.D., Capstone Project Advisor Date _________________________________________ _5/4/2015____________ Lawrence Ryan, Ph.D., Capstone Project Advisor Date _________________________________________ __5/4/2015___________ Don Lee, L.Ac., Specialty Chair Date _________________________________________ __5/4/2015____________ Andrea Murchison, DAOM, L.Ac., Program Director Date 2 Schulz Capstone ABSTRACT Research has shown that arresting progress of disease by early intervention is paramount to preventing its progression. Recent studies reveal early signs and symptoms of Parkinson’s Disease (PD) to be anosmia (loss of smell), constipation and REM Sleep Disorder and are confirmed to develop up to ten years prior to the more well known, and classical diagnosed, motor symptoms of resting tremor, bradykinesia, and rigidity. Motor symptoms in PD appear as a result of the progressive loss of dopamine in the basal ganglia, particularly within the substantia nigra, pars compacta region. By the time PD motor symptoms develop and are diagnosed, an estimated 80% of striatal nerve terminals and 60% of dopaminergic neurons have already been lost. Modern biomedical intervention for motor symptoms primarily focuses on the use of the pharmaceutical combination of carbidopa/levodopa (L-Dopa therapy). It is well known L- Dopa therapy has a waning period after 3 to 5 years of use, with up to 50% of patients developing dyskinesias as a result of this treatment. -
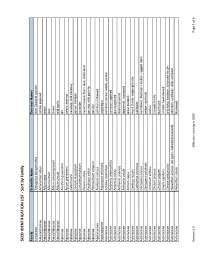
SEED IDENTIFICATION LIST - Sort by Family
SEED IDENTIFICATION LIST - Sort by Family Family Scientific Name Common Names Aizoaceae Tetragonia tetragonoides New Zealand spinach Amaranthaceae Amaranthus albus tumble pigweed Amaryllidaceae Allium cepa onion Amaryllidaceae Allium porrum leek Amaryllidaceae Allium schoenoprasum chives Amaryllidaceae Allium vineale wild garlic Apiaceae Anethum graveolens dill Apiaceae Apium graveolens celery, celeriac Apiaceae Carum carvi caraway; wild caraway Apiaceae Conium maculatum poison hemlock Apiaceae Coriandrum sativum coriander Apiaceae Daucus carota carrot; Queen Ane's lace; wild carrot Apiaceae Pastinaca sativa parsnip; wild parsnip Apiaceae Petroselinum crispum parsley Apocynaceae Asclepias syriaca common milkweed Asparagaceae Asparagus officinalis asparagus Asteraceae Achillea millefolium common yarrow, woolly yarrow Asteraceae Ambrosia artemisiifolia common ragweed Asteraceae Ambrosia trifida giant ragweed Asteraceae Anthemis arvensis field chamomile Asteraceae Anthemis cotula dogfennel, mayweed Asteraceae Arctium lappa great burdock Asteraceae Carduus nutans musk thistle, nodding thistle Asteraceae Carthamus tinctorius safflower Asteraceae Centaurea cyanus cornflower, bachelor's button, ragged robin Asteraceae Centaurea solstitialis yellow starthistle Asteraceae Cichorium endivia endive Asteraceae Cirsium arvense Canada thistle Asteraceae Cirsium vulgare bull thistle Asteraceae Crepis capillaris smooth hawksbeard Asteraceae Cynara cardunculus artichoke, cardoon, artichoke thistle Asteraceae Helianthus annuus (all types, cultivated and -

The Impact of the Flower Mite Aceria Acroptiloni on the Invasive Plant
BioControl (2014) 59:367–375 DOI 10.1007/s10526-014-9573-z The impact of the flower mite Aceria acroptiloni on the invasive plant Russian knapweed, Rhaponticum repens, in its native range Ghorbanali Asadi • Reza Ghorbani • Massimo Cristofaro • Philipp Chetverikov • Radmila Petanovic´ • Biljana Vidovic´ • Urs Schaffner Received: 26 October 2013 / Accepted: 13 March 2014 / Published online: 27 March 2014 Ó International Organization for Biological Control (IOBC) 2014 Abstract Rhaponticum repens (L.) Hidalgo is a clonal field site revealed that A. acroptiloni was by far the Asteraceae plant native to Asia and highly invasive in dominant mite species. We conclude that the mite A. North America. We conducted open-field experiments in acroptiloni is a promising biological control candidate Iran to assess the impact of the biological control inflicting significant impact on the above-ground biomass candidate, Aceria acroptiloni Shevchenko & Kovalev and reproductive output of the invasive plant R. repens. (Acari, Eriophyidae), on the target weed. Using three different experimental approaches, we found that mite Keywords Above-ground biomass Á Acari Á attack reduced the biomass of R. repens shoots by Acroptilon Á Asteraceae Á Classical biological 40–75 %. Except for the initial year of artificial infesta- control Á Pre-release studies Á Seed production tion by A. acroptiloni of R. repens shoots, the number of seed heads was reduced by 60–80 % and the number of seeds by 95–98 %. Morphological investigations of the Introduction mite complex attacking R. repens at the experimental The aim of pre-release studies in classical biological weed control projects is not only to experimentally Handling editor: John Scott. -

Analysis of the Trends in Biochemical Research Using Latent Dirichlet Allocation (LDA)
Article Analysis of the Trends in Biochemical Research Using Latent Dirichlet Allocation (LDA) Hee Jay Kang 1, Changhee Kim 1,* and Kyungtae Kang 2,* 1 College of Business Administration, Incheon National University, 119, Academy-ro, Yeonsu-gu, Incheon 22012, Korea; [email protected] 2 Department of Applied Chemistry, Kyung Hee University, 1732, Deogyeong-daero, Giheung-gu, Yongin-si, Gyeonggi-do 130-701, Korea * Correspondence: [email protected] (C.K.); [email protected] (K.K.); Tel.: +82-2-880-8594(C.K.) Received: 15 April 2019; Accepted: 13 June 2019; Published: 18 June 2019 Abstract: Biochemistry has been broadly defined as “chemistry of molecules included or related to living systems”, but is becoming increasingly hard to be distinguished from other related fields. Targets of its studies evolve rapidly; some newly emerge, disappear, combine, or resurface themselves with a fresh viewpoint. Methodologies for biochemistry have been extremely diversified, thanks particularly to those adopted from molecular biology, synthetic chemistry, and biophysics. Therefore, this paper adopts topic modeling, a text mining technique, to identify the research topics in the field of biochemistry over the past twenty years and quantitatively analyze the changes in its trends. The results of the topic modeling analysis obtained through this study will provide a helpful tool for researchers, journal editors, publishers, and funding agencies to understand the connections among the diverse sub-fields in biochemical research and even see how the research topics branch out and integrate with other fields. Keywords: biochemistry; topic modeling; research trend; LDA 1. Introduction Biochemistry is the study of the structure, composition, and chemical reactions of substances in living systems and includes the sciences of molecular biology, immunochemistry, and neurochemistry, as well as bioinorganic, bioorganic, and biophysical chemistry [1]. -

Neurochemical Mechanisms Underlying Alcohol Withdrawal
Neurochemical Mechanisms Underlying Alcohol Withdrawal John Littleton, MD, Ph.D. More than 50 years ago, C.K. Himmelsbach first suggested that physiological mechanisms responsible for maintaining a stable state of equilibrium (i.e., homeostasis) in the patient’s body and brain are responsible for drug tolerance and the drug withdrawal syndrome. In the latter case, he suggested that the absence of the drug leaves these same homeostatic mechanisms exposed, leading to the withdrawal syndrome. This theory provides the framework for a majority of neurochemical investigations of the adaptations that occur in alcohol dependence and how these adaptations may precipitate withdrawal. This article examines the Himmelsbach theory and its application to alcohol withdrawal; reviews the animal models being used to study withdrawal; and looks at the postulated neuroadaptations in three systems—the gamma-aminobutyric acid (GABA) neurotransmitter system, the glutamate neurotransmitter system, and the calcium channel system that regulates various processes inside neurons. The role of these neuroadaptations in withdrawal and the clinical implications of this research also are considered. KEY WORDS: AOD withdrawal syndrome; neurochemistry; biochemical mechanism; AOD tolerance; brain; homeostasis; biological AOD dependence; biological AOD use; disorder theory; biological adaptation; animal model; GABA receptors; glutamate receptors; calcium channel; proteins; detoxification; brain damage; disease severity; AODD (alcohol and other drug dependence) relapse; literature review uring the past 25 years research- science models used to study with- of the reasons why advances in basic ers have made rapid progress drawal neurochemistry as well as a research have not yet been translated Din understanding the chemi- reluctance on the part of clinicians to into therapeutic gains and suggests cal activities that occur in the nervous consider new treatments. -

ISTA List of Stabilized Plant Names 7Th Edition
ISTA List of Stabilized Plant Names th 7 Edition ISTA Nomenclature Committee Chair: Dr. M. Schori Published by All rights reserved. No part of this publication may be The Internation Seed Testing Association (ISTA) reproduced, stored in any retrieval system or transmitted Zürichstr. 50, CH-8303 Bassersdorf, Switzerland in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without prior ©2020 International Seed Testing Association (ISTA) permission in writing from ISTA. ISBN 978-3-906549-77-4 ISTA List of Stabilized Plant Names 1st Edition 1966 ISTA Nomenclature Committee Chair: Prof P. A. Linehan 2nd Edition 1983 ISTA Nomenclature Committee Chair: Dr. H. Pirson 3rd Edition 1988 ISTA Nomenclature Committee Chair: Dr. W. A. Brandenburg 4th Edition 2001 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 5th Edition 2007 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 6th Edition 2013 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 7th Edition 2019 ISTA Nomenclature Committee Chair: Dr. M. Schori 2 7th Edition ISTA List of Stabilized Plant Names Content Preface .......................................................................................................................................................... 4 Acknowledgements ....................................................................................................................................... 6 Symbols and Abbreviations .......................................................................................................................... -

Alien Plant Species in the Agricultural Habitats of Ukraine: Diversity and Risk Assessment
Ekológia (Bratislava) Vol. 37, No. 1, p. 24–31, 2018 DOI:10.2478/eko-2018-0003 ALIEN PLANT SPECIES IN THE AGRICULTURAL HABITATS OF UKRAINE: DIVERSITY AND RISK ASSESSMENT RAISA BURDA Institute for Evolutionary Ecology, NAS of Ukraine, 37, Lebedeva Str., 03143 Kyiv, Ukraine; e-mail: [email protected] Abstract Burda R.: Alien plant species in the agricultural habitats of Ukraine: diversity and risk assessment. Ekológia (Bratislava), Vol. 37, No. 1, p. 24–31, 2018. This paper is the first critical review of the diversity of the Ukrainian adventive flora, which has spread in agricultural habitats in the 21st century. The author’s annotated checklist con- tains the data on 740 species, subspecies and hybrids from 362 genera and 79 families of non-native weeds. The floristic comparative method was used, and the information was gen- eralised into some categories of five characteristic features: climamorphotype (life form), time and method of introduction, level of naturalisation, and distribution into 22 classes of three habitat types according to European Nature Information System (EUNIS). Two assess- ments of the ecological risk of alien plants were first conducted in Ukraine according to the European methods: the risk of overcoming natural migration barriers and the risk of their impact on the environment. The exposed impact of invasive alien plants on ecosystems has a convertible character; the obtained information confirms a high level of phytobiotic contami- nation of agricultural habitats in Ukraine. It is necessary to implement European and national documents regarding the legislative and regulative policy on invasive alien species as one of the threats to biotic diversity.