MTO1 Mediates Tissue Specificity of OXPHOS Defects Via Trna
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Role of Mitochondrial Ribosomal Protein S18-2 in Cancerogenesis and in Regulation of Stemness and Differentiation
From THE DEPARTMENT OF MICROBIOLOGY TUMOR AND CELL BIOLOGY (MTC) Karolinska Institutet, Stockholm, Sweden ROLE OF MITOCHONDRIAL RIBOSOMAL PROTEIN S18-2 IN CANCEROGENESIS AND IN REGULATION OF STEMNESS AND DIFFERENTIATION Muhammad Mushtaq Stockholm 2017 All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by E-Print AB 2017 © Muhammad Mushtaq, 2017 ISBN 978-91-7676-697-2 Role of Mitochondrial Ribosomal Protein S18-2 in Cancerogenesis and in Regulation of Stemness and Differentiation THESIS FOR DOCTORAL DEGREE (Ph.D.) By Muhammad Mushtaq Principal Supervisor: Faculty Opponent: Associate Professor Elena Kashuba Professor Pramod Kumar Srivastava Karolinska Institutet University of Connecticut Department of Microbiology Tumor and Cell Center for Immunotherapy of Cancer and Biology (MTC) Infectious Diseases Co-supervisor(s): Examination Board: Professor Sonia Lain Professor Ola Söderberg Karolinska Institutet Uppsala University Department of Microbiology Tumor and Cell Department of Immunology, Genetics and Biology (MTC) Pathology (IGP) Professor George Klein Professor Boris Zhivotovsky Karolinska Institutet Karolinska Institutet Department of Microbiology Tumor and Cell Institute of Environmental Medicine (IMM) Biology (MTC) Professor Lars-Gunnar Larsson Karolinska Institutet Department of Microbiology Tumor and Cell Biology (MTC) Dedicated to my parents ABSTRACT Mitochondria carry their own ribosomes (mitoribosomes) for the translation of mRNA encoded by mitochondrial DNA. The architecture of mitoribosomes is mainly composed of mitochondrial ribosomal proteins (MRPs), which are encoded by nuclear genomic DNA. Emerging experimental evidences reveal that several MRPs are multifunctional and they exhibit important extra-mitochondrial functions, such as involvement in apoptosis, protein biosynthesis and signal transduction. Dysregulations of the MRPs are associated with severe pathological conditions, including cancer. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

TRMT2B Is Responsible for Both Trna and Rrna M5u-Methylation in Human Mitochondria
ManuscriptbioRxiv - with preprint full author doi: https://doi.org/10.1101/797472 details ; this version posted October 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. TRMT2B is responsible for both tRNA and rRNA m5U-methylation in human mitochondria 1 1* Christopher A. Powell and Michal Minczuk 1 Medical Research Council Mitochondrial Biology Unit, University of Cambridge, Hills Road, Cambridge CB2 0XY, UK *Corresponding author: Michal Minczuk, e-mail: [email protected] Medical Research Council Mitochondrial Biology Unit, University of Cambridge, The Keith Peters Building, Cambridge Biomedical Campus, Hills Road, Cambridge, CB2 0XY, UK Phone: +44 (0)1223 252750 1 bioRxiv preprint doi: https://doi.org/10.1101/797472; this version posted October 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract RNA species play host to a plethora of post-transcriptional modifications which together make up the epitranscriptome. 5-methyluridine (m5U) is one of the most common modifications made to cellular RNA, where it is found almost ubiquitously in bacterial and eukaryotic cytosolic tRNAs at position 54. Here, we demonstrate that m5U54 in human mitochondrial tRNAs is catalysed by the nuclear-encoded enzyme TRMT2B, and that its repertoire of substrates is expanded to ribosomal RNAs, catalysing m5U429 in 12S rRNA. We show that TRMT2B is not essential for viability in human cells and that knocking-out the gene shows no obvious phenotype with regards to RNA stability, mitochondrial translation, or cellular growth. -

Short-Term Western-Style Diet Negatively Impacts Reproductive Outcomes in Primates
Short-term Western-style diet negatively impacts reproductive outcomes in primates Sweta Ravisankar, … , Shawn L. Chavez, Jon D. Hennebold JCI Insight. 2021;6(4):e138312. https://doi.org/10.1172/jci.insight.138312. Research Article Metabolism Reproductive biology A maternal Western-style diet (WSD) is associated with poor reproductive outcomes, but whether this is from the diet itself or underlying metabolic dysfunction is unknown. Here, we performed a longitudinal study using regularly cycling female rhesus macaques (n = 10) that underwent 2 consecutive in vitro fertilization (IVF) cycles, one while consuming a low-fat diet and another 6–8 months after consuming a high-fat WSD. Metabolic data were collected from the females prior to each IVF cycle. Follicular fluid (FF) and oocytes were assessed for cytokine/steroid levels and IVF potential, respectively. Although transition to a WSD led to weight gain and increased body fat, no difference in insulin levels was observed. A significant decrease in IL-1RA concentration and the ratio of cortisol/cortisone was detected in FF after WSD intake. Despite an increased probability of isolating mature oocytes, a 44% reduction in blastocyst number was observed with WSD consumption, and time-lapse imaging revealed delayed mitotic timing and multipolar divisions. RNA sequencing of blastocysts demonstrated dysregulation of genes involved in RNA binding, protein channel activity, mitochondrial function and pluripotency versus cell differentiation after WSD consumption. Thus, short-term WSD consumption promotes a proinflammatory intrafollicular microenvironment that is associated with impaired preimplantation development in the absence of large-scale metabolic changes. Find the latest version: https://jci.me/138312/pdf RESEARCH ARTICLE Short-term Western-style diet negatively impacts reproductive outcomes in primates Sweta Ravisankar,1,2 Alison Y. -

Lncrna SNHG8 Is Identified As a Key Regulator of Acute Myocardial
Zhuo et al. Lipids in Health and Disease (2019) 18:201 https://doi.org/10.1186/s12944-019-1142-0 RESEARCH Open Access LncRNA SNHG8 is identified as a key regulator of acute myocardial infarction by RNA-seq analysis Liu-An Zhuo, Yi-Tao Wen, Yong Wang, Zhi-Fang Liang, Gang Wu, Mei-Dan Nong and Liu Miao* Abstract Background: Long noncoding RNAs (lncRNAs) are involved in numerous physiological functions. However, their mechanisms in acute myocardial infarction (AMI) are not well understood. Methods: We performed an RNA-seq analysis to explore the molecular mechanism of AMI by constructing a lncRNA-miRNA-mRNA axis based on the ceRNA hypothesis. The target microRNA data were used to design a global AMI triple network. Thereafter, a functional enrichment analysis and clustering topological analyses were conducted by using the triple network. The expression of lncRNA SNHG8, SOCS3 and ICAM1 was measured by qRT-PCR. The prognostic values of lncRNA SNHG8, SOCS3 and ICAM1 were evaluated using a receiver operating characteristic (ROC) curve. Results: An AMI lncRNA-miRNA-mRNA network was constructed that included two mRNAs, one miRNA and one lncRNA. After RT-PCR validation of lncRNA SNHG8, SOCS3 and ICAM1 between the AMI and normal samples, only lncRNA SNHG8 had significant diagnostic value for further analysis. The ROC curve showed that SNHG8 presented an AUC of 0.850, while the AUC of SOCS3 was 0.633 and that of ICAM1 was 0.594. After a pairwise comparison, we found that SNHG8 was statistically significant (P SNHG8-ICAM1 = 0.002; P SNHG8-SOCS3 = 0.031). -

Supplementary Dataset S2
mitochondrial translational termination MRPL28 MRPS26 6 MRPS21 PTCD3 MTRF1L 4 MRPL50 MRPS18A MRPS17 2 MRPL20 MRPL52 0 MRPL17 MRPS33 MRPS15 −2 MRPL45 MRPL30 MRPS27 AURKAIP1 MRPL18 MRPL3 MRPS6 MRPS18B MRPL41 MRPS2 MRPL34 GADD45GIP1 ERAL1 MRPL37 MRPS10 MRPL42 MRPL19 MRPS35 MRPL9 MRPL24 MRPS5 MRPL44 MRPS23 MRPS25 ITB ITB ITB ITB ICa ICr ITL original ICr ICa ITL ICa ITL original ICr ITL ICr ICa mitochondrial translational elongation MRPL28 MRPS26 6 MRPS21 PTCD3 MRPS18A 4 MRPS17 MRPL20 2 MRPS15 MRPL45 MRPL52 0 MRPS33 MRPL30 −2 MRPS27 AURKAIP1 MRPS10 MRPL42 MRPL19 MRPL18 MRPL3 MRPS6 MRPL24 MRPS35 MRPL9 MRPS18B MRPL41 MRPS2 MRPL34 MRPS5 MRPL44 MRPS23 MRPS25 MRPL50 MRPL17 GADD45GIP1 ERAL1 MRPL37 ITB ITB ITB ITB ICa ICr original ICr ITL ICa ITL ICa ITL original ICr ITL ICr ICa translational termination MRPL28 MRPS26 6 MRPS21 PTCD3 C12orf65 4 MTRF1L MRPL50 MRPS18A 2 MRPS17 MRPL20 0 MRPL52 MRPL17 MRPS33 −2 MRPS15 MRPL45 MRPL30 MRPS27 AURKAIP1 MRPL18 MRPL3 MRPS6 MRPS18B MRPL41 MRPS2 MRPL34 GADD45GIP1 ERAL1 MRPL37 MRPS10 MRPL42 MRPL19 MRPS35 MRPL9 MRPL24 MRPS5 MRPL44 MRPS23 MRPS25 ITB ITB ITB ITB ICa ICr original ICr ITL ICa ITL ICa ITL original ICr ITL ICr ICa translational elongation DIO2 MRPS18B MRPL41 6 MRPS2 MRPL34 GADD45GIP1 4 ERAL1 MRPL37 2 MRPS10 MRPL42 MRPL19 0 MRPL30 MRPS27 AURKAIP1 −2 MRPL18 MRPL3 MRPS6 MRPS35 MRPL9 EEF2K MRPL50 MRPS5 MRPL44 MRPS23 MRPS25 MRPL24 MRPS33 MRPL52 EIF5A2 MRPL17 SECISBP2 MRPS15 MRPL45 MRPS18A MRPS17 MRPL20 MRPL28 MRPS26 MRPS21 PTCD3 ITB ITB ITB ITB ICa ICr ICr ITL original ITL ICa ICa ITL ICr ICr ICa original -

C6orf203 Controls OXPHOS Function Through Modulation of Mitochondrial Protein Biosynthesis
bioRxiv preprint doi: https://doi.org/10.1101/704403; this version posted July 17, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. C6orf203 controls OXPHOS function through modulation of mitochondrial protein biosynthesis number of characters excluding Materials and Methods: 40,651 Sara Palacios-Zambrano1,2, Luis Vázquez-Fonseca1,2, Cristina González-Páramos1,2, Laura Mamblona1,2, Laura Sánchez-Caballero3, Leo Nijtmans3, Rafael Garesse1,2 and Miguel Angel Fernández-Moreno1,2,* 1 Departamento de Bioquímica, Instituto de Investigaciones Biomédicas “Alberto Sols” UAM CSIC and Centro de Investigación Biomédica en Red en Enfermedades Raras (CIBERER). Facultad de Medicina, Universidad Autónoma de Madrid. Madrid 28029, Spain. 2 Instituto de Investigación Sanitaria Hospital 12 de Octubre (imas12), Madrid 28041, Spain. 3 Department of Pediatrics, Radboud Center for Mitochondrial Medicine, Radboud University Medical Center, Nijmegen, The Netherlands. * To whom correspondence should be addressed. Tel:+34 91 497 31 29; Email: [email protected] Running title “C6orf203 controls mt-proteins synthesis” bioRxiv preprint doi: https://doi.org/10.1101/704403; this version posted July 17, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ABSTRACT Mitochondria are essential organelles present in the vast majority of eukaryotic cells. Their central function is to produce cellular energy through the OXPHOS system, and functional alterations provoke so-called mitochondrial OXPHOS diseases. It is estimated that several hundred mitochondrial proteins have unknown functions. Very recently, C6orf203 was described to participate in mitochondrial transcription under induced mitochondrial DNA depletion stress conditions. -

CLPP Coordinates Mitoribosomal Assembly Through the Regulation of ERAL1 Levels
Article CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels Karolina Szczepanowska1,2,†, Priyanka Maiti1,2,†, Alexandra Kukat1,2, Eduard Hofsetz1,2, Hendrik Nolte1,3, Katharina Senft1,2, Christina Becker1,2, Benedetta Ruzzenente4, Hue-Tran Hornig-Do1,5, Rolf Wibom6, Rudolf J Wiesner1,5, Marcus Krüger1,3 & Aleksandra Trifunovic1,2,* Abstract the elimination of specific proteins when they are no longer needed. The irreversible nature of this process demands rigorous selectivity Despite being one of the most studied proteases in bacteria, very in the choice of substrates and the degradation timing. In prokary- little is known about the role of ClpXP in mitochondria. We now otes, regulated protein degradation is governed by energy-dependent present evidence that mammalian CLPP has an essential role in proteases, of which ClpXP is probably the best studied as it contri- determining the rate of mitochondrial protein synthesis by regu- butes to a large fraction of protein turnover in bacteria (Sauer & lating the level of mitoribosome assembly. Through a proteomic Baker, 2011). ClpXP is a highly conserved proteasome-like machin- approach and the use of a catalytically inactive CLPP, we produced ery present in all bacterial species and is also found in the mitochon- the first comprehensive list of possible mammalian ClpXP dria and chloroplasts of eukaryotic cells (Sauer & Baker, 2011). In substrates involved in the regulation of mitochondrial translation, mammals, ClpXP protein substrates are recognized and unfolded by oxidative phosphorylation, and a number of metabolic pathways. the hexameric AAA+ protein CLPX that delivers them to the CLPP We further show that the defect in mitoribosomal assembly is a protease for degradation (Sauer & Baker, 2011). -

PRODUCT SPECIFICATION Prest Antigen MRPS35 Product
PrEST Antigen MRPS35 Product Datasheet PrEST Antigen PRODUCT SPECIFICATION Product Name PrEST Antigen MRPS35 Product Number APrEST80950 Gene Description mitochondrial ribosomal protein S35 Alternative Gene MDS023, MRPS28 Names Corresponding Anti-MRPS35 (HPA038513) Antibodies Description Recombinant protein fragment of Human MRPS35 Amino Acid Sequence Recombinant Protein Epitope Signature Tag (PrEST) antigen sequence: DMEEYIWENSSSERNILETLLQMKAAEKNMEINKEELLGTKEIEEYKKSV VSLKNEEENENSISQYKESVK Fusion Tag N-terminal His6ABP (ABP = Albumin Binding Protein derived from Streptococcal Protein G) Expression Host E. coli Purification IMAC purification Predicted MW 26 kDa including tags Usage Suitable as control in WB and preadsorption assays using indicated corresponding antibodies. Purity >80% by SDS-PAGE and Coomassie blue staining Buffer PBS and 1M Urea, pH 7.4. Unit Size 100 µl Concentration Lot dependent Storage Upon delivery store at -20°C. Avoid repeated freeze/thaw cycles. Notes Gently mix before use. Optimal concentrations and conditions for each application should be determined by the user. Product of Sweden. For research use only. Not intended for pharmaceutical development, diagnostic, therapeutic or any in vivo use. No products from Atlas Antibodies may be resold, modified for resale or used to manufacture commercial products without prior written approval from Atlas Antibodies AB. Warranty: The products supplied by Atlas Antibodies are warranted to meet stated product specifications and to conform to label descriptions when used and stored properly. Unless otherwise stated, this warranty is limited to one year from date of sales for products used, handled and stored according to Atlas Antibodies AB's instructions. Atlas Antibodies AB's sole liability is limited to replacement of the product or refund of the purchase price. -

Transcriptomic and Proteomic Landscape of Mitochondrial
TOOLS AND RESOURCES Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals Inge Ku¨ hl1,2†*, Maria Miranda1†, Ilian Atanassov3, Irina Kuznetsova4,5, Yvonne Hinze3, Arnaud Mourier6, Aleksandra Filipovska4,5, Nils-Go¨ ran Larsson1,7* 1Department of Mitochondrial Biology, Max Planck Institute for Biology of Ageing, Cologne, Germany; 2Department of Cell Biology, Institute of Integrative Biology of the Cell (I2BC) UMR9198, CEA, CNRS, Univ. Paris-Sud, Universite´ Paris-Saclay, Gif- sur-Yvette, France; 3Proteomics Core Facility, Max Planck Institute for Biology of Ageing, Cologne, Germany; 4Harry Perkins Institute of Medical Research, The University of Western Australia, Nedlands, Australia; 5School of Molecular Sciences, The University of Western Australia, Crawley, Australia; 6The Centre National de la Recherche Scientifique, Institut de Biochimie et Ge´ne´tique Cellulaires, Universite´ de Bordeaux, Bordeaux, France; 7Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden Abstract Dysfunction of the oxidative phosphorylation (OXPHOS) system is a major cause of human disease and the cellular consequences are highly complex. Here, we present comparative *For correspondence: analyses of mitochondrial proteomes, cellular transcriptomes and targeted metabolomics of five [email protected] knockout mouse strains deficient in essential factors required for mitochondrial DNA gene (IKu¨ ); expression, leading to OXPHOS dysfunction. Moreover, -

Distinct Genetic Basis of Closely Related Toad Tadpoles Respectively Adapted to High Altitude and Karst Caves
G C A T T A C G G C A T genes Article Plateau Grass and Greenhouse Flower? Distinct Genetic Basis of Closely Related Toad Tadpoles Respectively Adapted to High Altitude and Karst Caves 1,2, 1, 1,2 1,2 1, Liming Chang y, Wei Zhu y, Shengchao Shi , Meihua Zhang , Jianping Jiang *, Cheng Li 1, Feng Xie 1 and Bin Wang 1,* 1 CAS Key Laboratory of Mountain Ecological Restoration and Bioresource Utilization & Ecological Restoration and Biodiversity Conservation Key Laboratory of Sichuan Province, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, China; [email protected] (L.C.); [email protected] (W.Z.); [email protected] (S.S.); [email protected] (M.Z.); [email protected] (C.L.); [email protected] (F.X.) 2 University of Chinese Academy of Sciences, Beijing 100049, China * Correspondence: [email protected] (B.W.); [email protected] (J.J.) These authors have contributed equally to this work. y Received: 1 December 2019; Accepted: 19 January 2020; Published: 22 January 2020 Abstract: Genetic adaptation to extremes is a fascinating topic. Nevertheless, few studies have explored the genetic adaptation of closely related species respectively inhabiting distinct extremes. With deep transcriptome sequencing, we attempt to detect the genetic architectures of tadpoles of five closely related toad species adapted to the Tibetan Plateau, middle-altitude mountains and karst caves. Molecular evolution analyses indicated that not only the number of fast evolving genes (FEGs), but also the functioning coverage of FEGs, increased with elevation. Enrichment analyses correspondingly revealed that the highland species had most of the FEGs involved in high-elevation adaptation, for example, amino acid substitutions of XRCC6 in its binding domains might improve the capacity of DNA repair of the toad. -
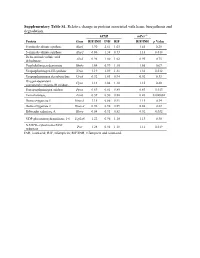
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator-