Biosimilars Event Summary
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

China's Health System Reform and Global Health Strategy in The
Testimony China’s Health System Reform and Global Health Strategy in the Context of COVID-19 Jennifer Bouey CT-A321-1 Testimony presented before the U.S.-China Economic and Security Review Commission on May 7, 2020. C O R P O R A T I O N For more information on this publication, visit www.rand.org/pubs/testimonies/CTA321-1.html Testimonies RAND testimonies record testimony presented or submitted by RAND associates to federal, state, or local legislative committees; government-appointed commissions and panels; and private review and oversight bodies. ChapterTitle 1 Published by the RAND Corporation, Santa Monica, Calif. © Copyright 2020 RAND Corporation RA® is a registered trademark. Limited Print and Electronic Distribution Rights This document and trademark(s) contained herein are protected by law. This representation of RAND intellectual property is provided for noncommercial use only. Unauthorized posting of this publication online is prohibited. Permission is given to duplicate this document for personal use only, as long as it is unaltered and complete. Permission is required from RAND to reproduce, or reuse in another form, any of its research documents for commercial use. For information on reprint and linking permissions, please visit www.rand.org/pubs/permissions.html. www.rand.org China’s Health System Reform and Global Health Strategy in the Context of COVID-19 Testimony of Jennifer Bouey1 The RAND Corporation2 Before the U.S.-China Economic and Security Review Commission May 7, 2020 hairman Cleveland, Commissioner Lee, and members of the Commission, thank you for inviting me to assess China’s pandemic-related issues regarding its public health C system, health care system, and global health strategy in the context of COVID-19. -

Expensive Patients, Reinsurance, and the Future of Health Care Reform
Emory Law Journal Volume 69 Issue 6 2020 Expensive Patients, Reinsurance, and the Future of Health Care Reform Govind Persad Follow this and additional works at: https://scholarlycommons.law.emory.edu/elj Recommended Citation Govind Persad, Expensive Patients, Reinsurance, and the Future of Health Care Reform, 69 Emory L. J. 1153 (2020). Available at: https://scholarlycommons.law.emory.edu/elj/vol69/iss6/1 This Article is brought to you for free and open access by the Journals at Emory Law Scholarly Commons. It has been accepted for inclusion in Emory Law Journal by an authorized editor of Emory Law Scholarly Commons. For more information, please contact [email protected]. PERSAD_8.21.20 8/24/2020 11:43 AM EXPENSIVE PATIENTS, REINSURANCE, AND THE FUTURE OF HEALTH CARE REFORM Govind Persad* ABSTRACT In 2017, Americans spent over $3.4 trillion—nearly 18% of gross domestic product—on health care. This spending is unevenly distributed: Almost a quarter is spent on the costliest 1% of patients, and almost half on the costliest 5%. Most of these patients soon return to a lower percentile, but many continue to incur health care costs in the top percentiles year after year. This Article focuses on the challenges that persistently expensive patients present for health law and policy, and how fairly dividing their medical costs among payers illuminates fundamental normative choices about the design and reform of health insurance. In doing so, this Article draws on bioethical and health policy analyses of the fair distribution of medical costs, and examines how legal doctrine shapes health systems’ options for responding to expensive patients. -

Addressing Health Care Market Consolidation and High Prices the Role of the States
HEALTH POLICY CENTER RESEARCH REPORT Addressing Health Care Market Consolidation and High Prices The Role of the States Robert A. Berenson Jaime S. King Katherine L. Gudiksen Roslyn Murray URBAN INSTITUTE UC HASTINGS LAW UC HASTINGS LAW GEORGETOWN UNIVERSITY Adele Shartzer URBAN INSTITUTE January 2020 ABOUT THE URBAN INSTITUTE The nonprofit Urban Institute is a leading research organization dedicated to developing evidence-based insights that improve people’s lives and strengthen communities. For 50 years, Urban has been the trusted source for rigorous analysis of complex social and economic issues; strategic advice to policymakers, philanthropists, and practitioners; and new, promising ideas that expand opportunities for all. Our work inspires effective decisions that advance fairness and enhance the well-being of people and places. Copyright © January 2020. Urban Institute. Permission is granted for reproduction of this file, with attribution to the Urban Institute. Cover image by Tim Meko. Contents Acknowledgments iv Executive Summary v 1. Introduction 1 2. Employee Retirement Income Security’s Act 8 3. State Efforts to Promote Transparency 11 4. State Efforts to Regulate Consolidation and Promote Competition 17 5. State Options for Overseeing and Regulating Prices 44 6. Conclusion 68 Notes 69 References 79 About the Authors 86 Statement of Independence 87 Acknowledgments This report was funded by the Commonwealth Fund. We are grateful to them and to all our funders, who make it possible for Urban to advance its mission. The views expressed are those of the authors and should not be attributed to the Urban Institute or UC Hastings Law, its trustees, or its funders. -
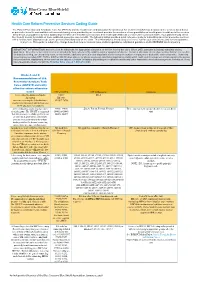
Health Care Reform Preventive Services Coding Guide
Health Care Reform Preventive Services Coding Guide The Patient Protection and Affordable Care Act (PPACA) and the Health Care and Education Reconciliation Act of 2010 (HCERA) has designated the services listed below as preventive benefits and available with no cost-sharing when provided by an in-network provider for members of non-grandfathered health plans. In addition to the services listed below, your patient may have additional preventive care benefits covered under their health plan that may or may not be covered at 100%. Your patient should check their benefit booklet for details on these additional preventive care benefits. The following tables provide a quick reference guide for submitting claims for preventive services with a “well-person” diagnosis code as the primary (first) diagnosis on the claim. This information is intended as a reference tool for your convenience and is not a guarantee of payment. This guide is subject to change based on new or revised laws and/or regulations, additional guidance and/or BCBSNC medical policy. IMPORTANT INFORMATION: Services must be billed with the appropriate diagnosis, at the line level of the claim (Block 24E), pursuant to industry standard coding guidelines. Preventive or screening services are intended for those who currently exhibit no signs or symptoms of disease. Services otherwise deemed preventive that are received in an inpatient setting, an emergency room, or that include additional procedures or diagnostic services may be subject to copayment, deductible and coinsurance. Submitting screening service codes (CPT, HCPCs, ICD-9 or ICD-10) when signs or symptoms are present constitutes inappropriate coding which could result in recoupment of monies paid to the provider for those services. -

Big Bang Health Care Reform — Does It Work?: the Case of Britain’S 1991 National Health Service Reforms
Big Bang Health Care Reform — Does It Work?: The Case of Britain’s 1991 National Health Service Reforms RUDOLF KLEIN University o f Bath, England ealth care reform has b e e n o n e of the worldwide epidemics of the 1990s. But among the many coun tries that have either attempted or contemplated the reform H of their health care systems (Hurst 1992; Organization for Economic Co-operation and Development 1994) Britain stands out from the rest. The reforms of the National Health Service (NHS) introduced in 1991 by Mrs. Thatcher’s Conservative government were driven by the much the same set of concerns and ideas that shaped the international vocabu lary of debate. In particular, they reflected the widely held belief that the best way of improving efficiency was to change the incentives to pro viders and that some form of marketlike competition was the best tool for achieving this aim (Saltman and van Otter 1992). In all these re spects, there was nothing all that special about Britain. What makes the British case special, and worthy of further study, however, is the ambi tious scope of the reforms and the relentless determination with which Mrs. Thatcher’s government implemented them. In the United States, the Clinton reforms plan foundered on the rocks of congressional oppo sition; in Sweden, a succession of local experiments and committees of inquiry failed to create a national consensus about the direction of health care policy; in the Netherlands, an ambitious plan of reform was The Milbank Quarterly, Vol. 73, No. 3, 1995 © 1995 Milbank Memorial Fund. -

Prevention Provisions in the Affordable Care Act Gail Shearer, MPP
American Public Health Association OCTOBER 2010 Prevention Provisions in the Affordable Care Act Gail Shearer, MPP Executive Summary n 2010 Congress enacted the Affordable Care Act, a historic and vigorously debated law designed to dramatically overhaul the health system. Included in the Affordable Care Act are comprehensive prevention provisions consistent with those called for by the American Public Health Association I (APHA) in its health reform agenda and supported by other leading experts in population health and prevention.12 The Affordable Care Act, if it is adequately funded, effectively implemented, and creatively leveraged through public and private-sector partnerships, will mark the turning point in the fundamental nature of our health system, initiating the transformation of our health system from one that treats sickness to one that promotes health and wellness. This issue brief begins (Section III) by summarizing the state of public health in the United States, including some measures of the growth of preventable diseases. Section IV describes the major provisions of the Affordable Care Act that address prevention through: (1) investing in public health; (2) educating the public; (3) expanding insurance coverage and requiring that health insur- ance include recommended preventive benefits; and (4) building capacity for better prevention in the future through demonstrations, research and evaluation. 800 I Street, NW • Washington, DC 20001-3710 • 202-777-APHA • fax: 202-777-2534 • www.apha.org Section V identifies key implementation -

The Health Care Reform Spectrum CHRO Education Series
I II III IV V VI I. II. I. Nearly a decade after the passage of the Affordable Care Act, health care continues to be a top issue among voters in the U.S., with the two major parties offering distinctly III. different visions of the direction of future reform. To help CHROs, their teams, stakeholders, and the public better understand the health care landscape and the prospects for change, we’re launching a series of short papers that will discuss the IV. various types of reform proposals being discussed among policy makers, political candidates and other key stakeholders. OBJECTIVES V. OF THE SERIES We hope this CHRO Education Series will VI. increase member awareness of the spectrum of potential future health care reforms that may be proposed over the next two years – and will help senior HR leaders assess the implications of various types of reform on their future business and talent strategies. We will also use these papers as the basis for follow up discussions with members in 2019 that will help shape the Association’s work with policymakers. 1 II. Policy Makers Have It’s also not surprising that Democrats and Differing Visions of Republicans have very different visions of what direction health care reform should Health Care Reform take in the future. While Republicans have largely abandoned prior calls to “repeal and The US health care system is unique among replace” the Affordable Care Act (ACA), industrialized nations in two ways: it does the Trump Administration declined to not provide universal coverage for all defend the ACA in a recent challenge in a citizens, and access to coverage for nearly Texas district court. -

Implementing Health Care Reform Policies in China: Challenges
a report of the csis freeman chair in china studies Implementing Health Care Reform Policies in China challenges and opportunities 1800 K Street, NW | Washington, DC 20006 Tel: (202) 887-0200 | Fax: (202) 775-3199 Editors E-mail: [email protected] | Web: www.csis.org Charles W. Freeman III Xiaoqing Lu Boynton December 2011 ISBN 978-0-89206-679-7 Ë|xHSKITCy066797zv*:+:!:+:! a report of the csis freeman chair in china studies Implementing Health Care Reform Policies in China challenges and opportunities Editors Charles W. Freeman III Xiaoqing Lu Boynton December 2011 About CSIS At a time of new global opportunities and challenges, the Center for Strategic and International Studies (CSIS) provides strategic insights and bipartisan policy solutions to decisionmakers in government, international institutions, the private sector, and civil society. A bipartisan, nonprofit organization headquartered in Washington, D.C., CSIS conducts research and analysis and devel- ops policy initiatives that look into the future and anticipate change. Founded by David M. Abshire and Admiral Arleigh Burke at the height of the Cold War, CSIS was dedicated to finding ways for America to sustain its prominence and prosperity as a force for good in the world. Since 1962, CSIS has grown to become one of the world’s preeminent international policy institutions, with more than 220 full-time staff and a large network of affiliated scholars focused on defense and security, regional stability, and transnational challenges ranging from energy and climate to global development and economic integration. Former U.S. senator Sam Nunn became chairman of the CSIS Board of Trustees in 1999, and John J. -

Principles for Health Care Reform
444 East Algonquin Road Arlington Heights, IL 60005-4664 ASPS POSITION STATEMENT 847-228-9900 www.plasticsurgery.org ASPS PRINCIPLES FOR HEALTH CARE REFORM While the ACA resulted in a number of desirable reforms, including coverage of pre-existing conditions, removal of lifetime caps, and coverage of children to age 26, the American health care system remains beset by a number of serious problems. Health insurance costs continue to rise, and private insurers draw ever-more-narrow networks, threatening broad access to care. Health system consolidation has exploded, threatening the viability of private practice and increasing costs to patients, taxpayers, and insurers. Well-meaning efforts to use alternative payment methods to reward the value of care, rather than the volume, have resulted in systems ill-equipped to integrate, analyze and reward specialist services. These systems threaten the viability of independent medical practices across the country, while causing physicians to retire early or sell their practices to large corporations and hospitals As the 115th Congress begins and a new administration takes power, policymakers are poised to again undertake health care reform. The following are key principles that the American Society of Plastic Surgeons believes must accompany such an effort. MAKE PATIENT ACCESS TO CARE PRIORITY ONE Patients should have timely access to high-quality medical care from the appropriate provider. To advance this goal, health care reform must - o Maximize the availability of high-quality, affordable coverage. o Prohibit denial or cancellation of coverage for pre-existing conditions. o Prohibit denial or cancellation of policies when a patient becomes sick. o Prohibit annual and lifetime caps. -

Health Care Reform: Tracking Tribal, Federal, and State Implementation
Executive Summary This report analyzes the impact of national health care reform on American Indian and Alaska Native (AI/AN) people. Twenty states are included in a systematic study of the most significant aspects of health care reform. The report identifies which elements of national health care reform are most likely to affect health care access for AI/ANs. It also projects the extent of these changes in health care access in the 20 study states under various scenarios. To provide this analysis, the report accesses data on Medicaid expenditures for AI/ANs provided by a series of studies sponsored by the Centers for Medicare & Medicaid Services (CMS) and conducted by the California Rural Indian Health Board (CRIHB), as well as health, demographic, and other data from the American Community Survey (ACS).1 Two elements of the Affordable Care Act of 2010 will likely have a substantial effect on AI/ANs: Medicaid expansion and health insurance exchanges, including exchange subsidies. As this report describes, Medicaid expansion will likely increase the number of AI/ANs enrolled in Medicaid in the 20 study states by an estimated 286,000 if aggressive outreach and enrollment practices are funded. Predicting participation in health insurance exchanges is more difficult, but an estimated 364,000 AI/ANs in the study states are likely eligible for exchange subsidies. Medicaid expansion, not without its own challenges, will be far easier to achieve than AI/AN participation in the health insurance exchanges. This report examines variations between the 20 study states along a number of salient indicators including measures described in the following table. -

Marketing and Promotion Facts
INTRODUCTION Activities conducted as part of pharmaceutical marketing and promotion are an important component of educating and informing consumers and health care professionals about new treatments. Direct-to-consumer (DTC) advertisements aim to inform patients of important treatment options, while pharmaceutical sales representatives work to get accurate, up-to- date information on medicines to health care professionals. These efforts have also been the subject of debate, with some questioning their value. This booklet offers facts about pharmaceutical marketing and promotion. We believe these facts are important to consider as the value of marketing and promotion are debated. Since our last publication on marketing and promotion,1 the pharmaceutical industry has worked to improve the dissemination of information about medical advances and to address concerns. One important change was the unanimous approval by PhRMA’s Board of Directors of Guiding Principles on Direct to Consumer Advertisements About Prescription Medicines. These voluntary Principles express the commitment of PhRMA members to deliver DTC communications that are a valuable contribution to public health. In addition, in 2008 PhRMA adopted a newly revised Code on Interactions with Health Care Professionals. The strengthened code refl ects a commitment to maintaining the highest ethical standards in all marketing practices and to promote the best patient care possible. This publication shows the role of marketing and promotion in speeding the dissemination of valuable improvements in medical care. It also highlights the important role that marketing plays in getting patients to discuss a range of health issues with their physicians, resulting in patients receiving needed treatment. We hope that the information contained in this booklet will enhance dialogue surrounding pharmaceutical marketing and promotion by providing a perspective that often is not heard. -

Lessons from 10 Years of Healthcare Reform in China
Practice BMJ Glob Health: first published as 10.1136/bmjgh-2019-002086 on 19 March 2020. Downloaded from Towards universal health coverage: lessons from 10 years of healthcare reform in China 1 1 2 3 4 Wenjuan Tao , Zhi Zeng, Haixia Dang, Bingqing Lu, Linh Chuong, Dahai Yue,4 Jin Wen,1 Rui Zhao,5 Weimin Li,6 Gerald F Kominski4,7 To cite: Tao W, Zeng Z, Dang H, ABSTRACT Summary box et al. Towards universal health Universal health coverage (UHC) is driving the global health coverage: lessons from 10 agenda. Many countries have embarked on national policy ► Continued political support is the most important years of healthcare reform reforms towards this goal, including China. In 2009, the in China. BMJ Global Health enabling condition for achieving universal health Chinese government launched a new round of healthcare 2020;5:e002086. doi:10.1136/ coverage (UHC). China has shown clear political will- reform towards UHC, aiming to provide universal coverage bmjgh-2019-002086 ingness to make UHC achievement a more country- of basic healthcare by the end of 2020. The year of led process. 2019 marks the 10th anniversary of China’s most recent Handling editor Seye Abimbola ► Increasing health financing is necessary, and the in- healthcare reform. Sharing China’s experience is especially vestment from both government and private sector timely for other countries pursuing reforms to achieve WT and ZZ contributed equally. is considered. UHC. This study describes the social, economic and health ► A strong primary healthcare system should be re- Received 16 October 2019 context in China, and then reviews the overall progress of garded as a core component in realising UHC.