A Competitive O-Acetylserine Sulfhydrylase Inhibitor Modulates the Formation of Cysteine Synthase Complex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Putative Cystathionine Beta-Synthase Homolog of Mycolicibacterium Smegmatis Is Involved in De Novo Cysteine Biosynthesis
University of Arkansas, Fayetteville ScholarWorks@UARK Theses and Dissertations 5-2020 A Putative Cystathionine Beta-Synthase Homolog of Mycolicibacterium smegmatis is Involved in de novo Cysteine Biosynthesis Saroj Kumar Mahato University of Arkansas, Fayetteville Follow this and additional works at: https://scholarworks.uark.edu/etd Part of the Cell Biology Commons, Molecular Biology Commons, and the Pathogenic Microbiology Commons Citation Mahato, S. K. (2020). A Putative Cystathionine Beta-Synthase Homolog of Mycolicibacterium smegmatis is Involved in de novo Cysteine Biosynthesis. Theses and Dissertations Retrieved from https://scholarworks.uark.edu/etd/3639 This Thesis is brought to you for free and open access by ScholarWorks@UARK. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of ScholarWorks@UARK. For more information, please contact [email protected]. A Putative Cystathionine Beta-Synthase Homolog of Mycolicibacterium smegmatis is Involved in de novo Cysteine Biosynthesis A thesis submitted in partial fulfillment of the requirement for the degree of Master of Science in Cell and Molecular Biology by Saroj Kumar Mahato Purbanchal University Bachelor of Science in Biotechnology, 2016 May 2020 University of Arkansas This thesis is approved for recommendation to the Graduate Council. ___________________________________ Young Min Kwon, Ph.D. Thesis Director ___________________________________ ___________________________________ Suresh Thallapuranam, Ph.D. Inés Pinto, Ph.D. Committee Member Committee Member ABSTRACT Mycobacteria include serious pathogens of humans and animals. Mycolicibacterium smegmatis is a non-pathogenic model that is widely used to study core mycobacterial metabolism. This thesis explores mycobacterial pathways of cysteine biosynthesis by generating and study of genetic mutants of M. smegmatis. Published in vitro biochemical studies had revealed three independent routes to cysteine synthesis in mycobacteria involving separate homologs of cysteine synthase, namely CysK1, CysK2, and CysM. -

Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma Divulgatum
microorganisms Article Proteome Cold-Shock Response in the Extremely Acidophilic Archaeon, Cuniculiplasma divulgatum Rafael Bargiela 1 , Karin Lanthaler 1,2, Colin M. Potter 1,2 , Manuel Ferrer 3 , Alexander F. Yakunin 1,2, Bela Paizs 1,2, Peter N. Golyshin 1,2 and Olga V. Golyshina 1,2,* 1 School of Natural Sciences, Bangor University, Deiniol Rd, Bangor LL57 2UW, UK; [email protected] (R.B.); [email protected] (K.L.); [email protected] (C.M.P.); [email protected] (A.F.Y.); [email protected] (B.P.); [email protected] (P.N.G.) 2 Centre for Environmental Biotechnology, Bangor University, Deiniol Rd, Bangor LL57 2UW, UK 3 Systems Biotechnology Group, Department of Applied Biocatalysis, CSIC—Institute of Catalysis, Marie Curie 2, 28049 Madrid, Spain; [email protected] * Correspondence: [email protected]; Tel.: +44-1248-388607; Fax: +44-1248-382569 Received: 27 April 2020; Accepted: 15 May 2020; Published: 19 May 2020 Abstract: The archaeon Cuniculiplasma divulgatum is ubiquitous in acidic environments with low-to-moderate temperatures. However, molecular mechanisms underlying its ability to thrive at lower temperatures remain unexplored. Using mass spectrometry (MS)-based proteomics, we analysed the effect of short-term (3 h) exposure to cold. The C. divulgatum genome encodes 2016 protein-coding genes, from which 819 proteins were identified in the cells grown under optimal conditions. In line with the peptidolytic lifestyle of C. divulgatum, its intracellular proteome revealed the abundance of proteases, ABC transporters and cytochrome C oxidase. From 747 quantifiable polypeptides, the levels of 582 proteins showed no change after the cold shock, whereas 104 proteins were upregulated suggesting that they might be contributing to cold adaptation. -

Supplemental Methods
Supplemental Methods: Sample Collection Duplicate surface samples were collected from the Amazon River plume aboard the R/V Knorr in June 2010 (4 52.71’N, 51 21.59’W) during a period of high river discharge. The collection site (Station 10, 4° 52.71’N, 51° 21.59’W; S = 21.0; T = 29.6°C), located ~ 500 Km to the north of the Amazon River mouth, was characterized by the presence of coastal diatoms in the top 8 m of the water column. Sampling was conducted between 0700 and 0900 local time by gently impeller pumping (modified Rule 1800 submersible sump pump) surface water through 10 m of tygon tubing (3 cm) to the ship's deck where it then flowed through a 156 µm mesh into 20 L carboys. In the lab, cells were partitioned into two size fractions by sequential filtration (using a Masterflex peristaltic pump) of the pre-filtered seawater through a 2.0 µm pore-size, 142 mm diameter polycarbonate (PCTE) membrane filter (Sterlitech Corporation, Kent, CWA) and a 0.22 µm pore-size, 142 mm diameter Supor membrane filter (Pall, Port Washington, NY). Metagenomic and non-selective metatranscriptomic analyses were conducted on both pore-size filters; poly(A)-selected (eukaryote-dominated) metatranscriptomic analyses were conducted only on the larger pore-size filter (2.0 µm pore-size). All filters were immediately submerged in RNAlater (Applied Biosystems, Austin, TX) in sterile 50 mL conical tubes, incubated at room temperature overnight and then stored at -80oC until extraction. Filtration and stabilization of each sample was completed within 30 min of water collection. -

Structure of Soybean Serine Acetyltransferase
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 288, NO. 51, pp. 36463–36472, December 20, 2013 Published in the U.S.A. Structure of Soybean Serine Acetyltransferase and Formation of the Cysteine Regulatory Complex as a Molecular Chaperone* Received for publication, October 14, 2013, and in revised form, November 4, 2013 Published, JBC Papers in Press, November 13, 2013, DOI 10.1074/jbc.M113.527143 Hankuil Yi‡1, Sanghamitra Dey§1, Sangaralingam Kumaran¶, Soon Goo Leeʈ, Hari B. Krishnan**, and Joseph M. Jezʈ2 From the ‡Department of Biological Sciences, Chungnam National University, 220 Gung-Dong, Yuseong-Gu, Daejeon 305-764,Korea, the §Department of Biological Sciences, Presidency University, Kolkata, West Bengal 700073, India, the ¶Council of Scientific and Industrial Research, Institute of Microbial Technology, Sector 39-A, Chandigarh 160036, India, the ʈDepartment of Biology, Washington University, St. Louis, Missouri 63130, and the **Plant Genetics Research Unit, United States Department of Agriculture- Agricultural Research Service, Department of Agronomy, University of Missouri, Columbia, Missouri 65211 Background: Serine acetyltransferase (SAT) catalyzes the limiting step in cysteine biosynthesis. Results: Analysis of soybean SAT provides insight into catalysis and protein-protein interactions. Downloaded from Conclusion: Key structural features are required for catalysis and formation of a stable macromolecular complex. Significance: A new role for protein complex formation in plant cysteine biosynthesis is proposed. Serine acetyltransferase (SAT) catalyzes the limiting reaction sis plays a central role in fixing inorganic sulfur from the envi- in plant and microbial biosynthesis of cysteine. In addition to its ronment into the metabolic precursor for cellular thiol-con- http://www.jbc.org/ enzymatic function, SAT forms a macromolecular complex with taining compounds (3–6). -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -
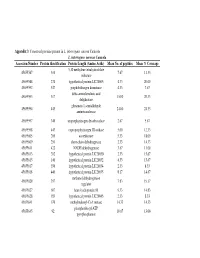
Appendix 3 and 4
Appendix 3 : Conserved proteins present in L. interrogans serovar Canicola L. interrogans serovar Canicola Accession Number Protein identification Protein Length (Amino Acids) Mean No. of peptides Mean % Coverage 5,10 methylene tetrahydrofolate 45655587 310 7.67 11.33 reductase 45655588 274 hypothetical protein LIC20005 4.33 20.00 45655592 547 porphobilinogen deaminase 4.33 7.67 delta-aminolevulinic acid 45655593 317 15.00 20.33 dehydratase glutamate-1-semialdehyde 45655594 443 24.00 24.33 aminotransferase 45655597 340 uroporphyrinogen decarboxylase 2.67 5.67 45655598 443 coproporphyrinogen III oxidase 5.00 12.33 45655605 200 azoreductase 5.33 18.00 45655609 251 short-chain dehydrogenase 2.33 14.33 45655611 422 NADH dehydrogenase 3.67 11.00 45655613 202 hypothetical protein LIC20030 2.33 15.67 45655615 140 hypothetical protein LIC20032 4.33 15.67 45655617 356 hypothetical protein LIC20034 2.33 8.33 45655618 440 hypothetical protein LIC20035 8.17 14.67 methanol dehydrogenase 45655620 357 7.83 19.17 regulator 45655627 607 heat shock protein 90 9.33 14.83 45655628 189 hypothetical protein LIC20045 2.33 8.33 45655641 670 methylmalonyl-CoA mutase 14.33 14.33 phosphoribosyl-ATP 45655645 92 10.67 13.00 pyrophosphatase 3-oxoacyl-(acyl-carrier protein) 45655647 254 4.00 17.00 reductase 45655648 77 acyl carrier protein 3.67 9.00 45655661 171 hypothetical protein LIC20078 5.00 26.00 4-hydroxybenzoyl-CoA 45655663 142 3.33 11.67 thioesterase S-adenosyl-L-homocysteine 45655666 436 13.00 19.67 hydrolase B12-dependent methionine 45655668 1247 42.67 21.67 -

Product Sheet Info
Product Information Sheet for NR-19703 Vibrio cholerae Gateway® Clone Set, Growth Conditions: Media: Recombinant in Escherichia coli, Plate 25 LB broth or agar containing 50 µg/mL kanamycin Incubation: Catalog No. NR-19703 Temperature: E. coli, strain DH10B-T1 clones should be This reagent is the tangible property of the U.S. Government. grown at 37°C. Atmosphere: Aerobic For research use only. Not for human use. Propagation: 1. Scrape top of frozen well with a pipette tip and streak onto Contributor: agar plate. Pathogen Functional Genomics Resource Center at the J. 2. Incubate the plates at 37°C for 1 day. Craig Venter Institute Citation: Manufacturer: Acknowledgment for publications should read “The following BEI Resources reagent was obtained through BEI Resources, NIAID, NIH: ® Vibrio cholerae Gateway Clone Set, Recombinant in Product Description: Escherichia coli, Plate 25, NR-19703.” Production in the 96-well format has increased risk of cross- contamination between adjacent wells. Individual clones Biosafety Level: 1 should be purified (e.g. single colony isolation and purification Appropriate safety procedures should always be used with this using good microbiological practices) and sequence-verified material. Laboratory safety is discussed in the following prior to use. BEI Resources does not confirm or validate publication: U.S. Department of Health and Human Services, individual mutants provided by the contributor. Public Health Service, Centers for Disease Control and Prevention, and National Institutes of Health. Biosafety in The Vibrio cholerae (V. cholerae) Gateway® clone set consists Microbiological and Biomedical Laboratories. 5th ed. of 46 plates which contain 3813 sequence validated clones Washington, DC: U.S. -

Granny Smith
www.nature.com/scientificreports OPEN Ethylene –dependent and –independent superfcial scald resistance mechanisms in ‘Granny Received: 19 March 2018 Accepted: 17 July 2018 Smith’ apple fruit Published: xx xx xxxx Evangelos Karagiannis1, Michail Michailidis1, Georgia Tanou1,2, Martina Samiotaki3, Katerina Karamanoli4, Evangelia Avramidou5, Ioannis Ganopoulos6, Panagiotis Madesis7 & Athanassios Molassiotis 1 Superfcial scald is a major physiological disorder of apple fruit (Malus domestica Borkh.) characterized by skin browning following cold storage; however, knowledge regarding the downstream processes that modulate scald phenomenon is unclear. To gain insight into the mechanisms underlying scald resistance, ‘Granny Smith’ apples after harvest were treated with diphenylamine (DPA) or 1-methylcyclopropene (1-MCP), then cold stored (0 °C for 3 months) and subsequently were ripened at room temperature (20 °C for 8 days). Phenotypic and physiological data indicated that both chemical treatments induced scald resistance while 1-MCP inhibited the ethylene-dependent ripening. A combination of multi-omic analysis in apple skin tissue enabled characterization of potential genes, proteins and metabolites that were regulated by DPA and 1-MCP at pro-symptomatic and scald- symptomatic period. Specifcally, we characterized strata of scald resistance responses, among which we focus on selected pathways including dehydroabietic acid biosynthesis and UDP-D-glucose regulation. Through this approach, we revealed scald-associated transcriptional, proteomic and metabolic signatures and identifed pathways modulated by the common or distinct functions of DPA and 1-MCP. Also, evidence is presented supporting that cytosine methylation-based epigenetic regulation is involved in scald resistance. Results allow a greater comprehension of the ethylene– dependent and –independent metabolic events controlling scald resistance. -
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Peter D Karp Ingrid Keseler An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Anamika Kothari Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000980815Cyc: Corynebacterium camporealensis DSM 44610 Cellular Overview Connections between pathways are omitted for legibility. Ron Caspi phosphate phosphate (S)-lactate phosphate ammonium predicted ABC RS04760 RS02955 RS06425 RS10630 transporter of phosphate phosphate (S)-lactate phosphate ammonium phosphate Amine and Tetrapyrrole Biosynthesis Amino Acid Degradation glutaminyl-tRNA gln Aminoacyl-tRNA Charging Polyamine a ring-opened 7- a DNA containing (1S,2R)-1- a [ThiI sulfur- biosynthesis via transamidation Biosynthesis an apo [peptidyl- all-trans- an L-asparaginyl- an L-cysteinyl- Polyprenyl Biosynthesis siroheme biosynthesis TCA cycle TCA cycle IV (2-oxoglutarate decarboxylase) L-valine degradation I L-asparagine methylguanine coenzyme A an apurinic/ ser C-(indol-3- carrier protein]- cys a [glutamine- L-isoleucine degradation I L-leucine degradation I L-threonine carrier protein] ATP retinyl palmitate [tRNA Asn ] [tRNA Cys ] dGDP spermidine degradation I in DNA apyrimidinic site yl)glycerol L-cysteine synthetase]- -

O O2 Enzymes Available from Sigma Enzymes Available from Sigma
COO 2.7.1.15 Ribokinase OXIDOREDUCTASES CONH2 COO 2.7.1.16 Ribulokinase 1.1.1.1 Alcohol dehydrogenase BLOOD GROUP + O O + O O 1.1.1.3 Homoserine dehydrogenase HYALURONIC ACID DERMATAN ALGINATES O-ANTIGENS STARCH GLYCOGEN CH COO N COO 2.7.1.17 Xylulokinase P GLYCOPROTEINS SUBSTANCES 2 OH N + COO 1.1.1.8 Glycerol-3-phosphate dehydrogenase Ribose -O - P - O - P - O- Adenosine(P) Ribose - O - P - O - P - O -Adenosine NICOTINATE 2.7.1.19 Phosphoribulokinase GANGLIOSIDES PEPTIDO- CH OH CH OH N 1 + COO 1.1.1.9 D-Xylulose reductase 2 2 NH .2.1 2.7.1.24 Dephospho-CoA kinase O CHITIN CHONDROITIN PECTIN INULIN CELLULOSE O O NH O O O O Ribose- P 2.4 N N RP 1.1.1.10 l-Xylulose reductase MUCINS GLYCAN 6.3.5.1 2.7.7.18 2.7.1.25 Adenylylsulfate kinase CH2OH HO Indoleacetate Indoxyl + 1.1.1.14 l-Iditol dehydrogenase L O O O Desamino-NAD Nicotinate- Quinolinate- A 2.7.1.28 Triokinase O O 1.1.1.132 HO (Auxin) NAD(P) 6.3.1.5 2.4.2.19 1.1.1.19 Glucuronate reductase CHOH - 2.4.1.68 CH3 OH OH OH nucleotide 2.7.1.30 Glycerol kinase Y - COO nucleotide 2.7.1.31 Glycerate kinase 1.1.1.21 Aldehyde reductase AcNH CHOH COO 6.3.2.7-10 2.4.1.69 O 1.2.3.7 2.4.2.19 R OPPT OH OH + 1.1.1.22 UDPglucose dehydrogenase 2.4.99.7 HO O OPPU HO 2.7.1.32 Choline kinase S CH2OH 6.3.2.13 OH OPPU CH HO CH2CH(NH3)COO HO CH CH NH HO CH2CH2NHCOCH3 CH O CH CH NHCOCH COO 1.1.1.23 Histidinol dehydrogenase OPC 2.4.1.17 3 2.4.1.29 CH CHO 2 2 2 3 2 2 3 O 2.7.1.33 Pantothenate kinase CH3CH NHAC OH OH OH LACTOSE 2 COO 1.1.1.25 Shikimate dehydrogenase A HO HO OPPG CH OH 2.7.1.34 Pantetheine kinase UDP- TDP-Rhamnose 2 NH NH NH NH N M 2.7.1.36 Mevalonate kinase 1.1.1.27 Lactate dehydrogenase HO COO- GDP- 2.4.1.21 O NH NH 4.1.1.28 2.3.1.5 2.1.1.4 1.1.1.29 Glycerate dehydrogenase C UDP-N-Ac-Muramate Iduronate OH 2.4.1.1 2.4.1.11 HO 5-Hydroxy- 5-Hydroxytryptamine N-Acetyl-serotonin N-Acetyl-5-O-methyl-serotonin Quinolinate 2.7.1.39 Homoserine kinase Mannuronate CH3 etc.