Betaxolol Hydrochloride
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Effect of Topical Beta-Adrenoceptor Blocking Agents on Pulsatile Ocular Blood Flow
THE EFFECT OF TOPICAL BETA-ADRENOCEPTOR BLOCKING AGENTS ON PULSATILE OCULAR BLOOD FLOW C. D. MORSMAN, M. E. BOSEM, M. LUSKY and R. N. WEINREB San Diego, California SUMMARY factors (e.g. hypertension, diabetes, peripheral Thirty-three ocular hypertensive patients (21 with vascular disease and vasospasm) to the vascular primary open angle glaucoma and 12 glaucoma system suggest that blood flow in the optic nerve suspects) were randomly assigned to receive either head and retina may be altered in glaucoma.4 Of the timolol, levobunolol or betaxolol in one eye. Pulsatile various vascular beds within the posterior segment, ocular blood flow (POBF) was measured before the choroidal circulation is of particular interest since treatment (baseline) and 2 hours after drop adminis it provides the major contribution to the blood flow tration. After 1 week of regular twice-daily dosage, of the optic nerve at the level of the lamina cribrosa.5 POBF was measured again both immediately before Beta-2 adrenergic receptors have been demon and 2 hours after drop instillation. All measurements strated in human optic nerve,6 as well as in choroidal were made by an investigator masked to treatment. and retinal blood vessels.7 Blockade of these POBF increased by 11% (p = 0.09) at week 0 after receptors can cause vasoconstrictionS which could levobunolol administration, and by 22% (p = 0.20) at adversely affect visual function if adequate concen week 1 before drop administration compared with trations of the drug diffused into the posterior baseline. It dropped by 23% and 25% (p = 0.04 and segment of the eye or were absorbed systemically. -

Brimonidine Tartrate; Brinzolamide
Contains Nonbinding Recommendations Draft Guidance on Brimonidine Tartrate ; Brinzolamide This draft guidance, when finalized, will represent the current thinking of the Food and Drug Administration (FDA, or the Agency) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. To discuss an alternative approach, contact the Office of Generic Drugs. Active Ingredient: Brimonidine tartrate; Brinzolamide Dosage Form; Route: Suspension/drops; ophthalmic Strength: 0.2%; 1% Recommended Studies: One study Type of study: Bioequivalence (BE) study with clinical endpoint Design: Randomized (1:1), double-masked, parallel, two-arm, in vivo Strength: 0.2%; 1% Subjects: Males and females with chronic open angle glaucoma or ocular hypertension in both eyes. Additional comments: Specific recommendations are provided below. ______________________________________________________________________________ Analytes to measure (in appropriate biological fluid): Not applicable Bioequivalence based on (95% CI): Clinical endpoint Additional comments regarding the BE study with clinical endpoint: 1. The Office of Generic Drugs (OGD) recommends conducting a BE study with a clinical endpoint in the treatment of open angle glaucoma and ocular hypertension comparing the test product to the reference listed drug (RLD), each applied as one drop in both eyes three times daily at approximately 8:00 a.m., 4:00 p.m., and 10:00 p.m. for 42 days (6 weeks). 2. Inclusion criteria (the sponsor may add additional criteria): a. Male or nonpregnant females aged at least 18 years with chronic open angle glaucoma or ocular hypertension in both eyes b. -

Use of Emulsions for Intra- and Periocular Injection
(19) & (11) EP 1 611 879 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61K 9/107 (2006.01) 12.08.2009 Bulletin 2009/33 (21) Application number: 04291684.1 (22) Date of filing: 02.07.2004 (54) Use of emulsions for intra- and periocular injection Verwendung von Emulsionen zur intra- und periocularen Injection Utilisation des émulsions pour injection intra- et périoculaire. (84) Designated Contracting States: (74) Representative: de Mareüil-Villette, Caroline et al AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Cabinet Plasseraud HU IE IT LI LU MC NL PL PT RO SE SI SK TR 52 rue de la Victoire 75440 Paris Cedex 09 (FR) (43) Date of publication of application: 04.01.2006 Bulletin 2006/01 (56) References cited: EP-A- 0 521 799 EP-A- 1 020 194 (73) Proprietors: WO-A-02/09667 WO-A-93/18852 • Novagali Pharma S.A. WO-A-94/05298 WO-A-03/053405 91000 Evry (FR) US-A- 5 632 984 • CENTRE NATIONAL DE LA RECHERCHE SCIENTIFIQUE (CNRS) • KLANG S H ET AL: "PHYSICOCHEMICAL 75016 Paris (FR) CHARACTERIAZATION AND ACUTE TOXICITY • INSTITUT NATIONAL DE LA SANTE ET DE LA EVALUATION OF A POSITIVELY-CHARGED RECHERCHE MEDICALE (INSERM) SUBMICRON EMULSION VEHICLE" JOURNAL 75013 Paris (FR) OF PHARMACY AND PHARMACOLOGY, • YISSUM RESEARCH DEVELOPMENT COMPANY LONDON, GB, vol. 46, no. 12, December 1994 OF THE HEBREW UNIVERSITY OF JERUSALEM (1994-12), pages 986-993, XP008005426 ISSN: 91390 Jerusalem (IL) 0022-3573 • KLANG S ET AL: "INFLUENCE OF EMULSION (72) Inventors: DROPLET SURFACE CHARGE ON • Rabinovich-Guilatt, Laura INDOMETHACIN OCULAR TISSUE 75015 Paris (FR) DISTRIBUTION" PHARMACEUTICAL • De Kozak, Yvonne DEVELOPMENT AND TECHNOLOGY, NEW 75013 Paris (FR) YORK, NY, US, vol. -

Title 16. Crimes and Offenses Chapter 13. Controlled Substances Article 1
TITLE 16. CRIMES AND OFFENSES CHAPTER 13. CONTROLLED SUBSTANCES ARTICLE 1. GENERAL PROVISIONS § 16-13-1. Drug related objects (a) As used in this Code section, the term: (1) "Controlled substance" shall have the same meaning as defined in Article 2 of this chapter, relating to controlled substances. For the purposes of this Code section, the term "controlled substance" shall include marijuana as defined by paragraph (16) of Code Section 16-13-21. (2) "Dangerous drug" shall have the same meaning as defined in Article 3 of this chapter, relating to dangerous drugs. (3) "Drug related object" means any machine, instrument, tool, equipment, contrivance, or device which an average person would reasonably conclude is intended to be used for one or more of the following purposes: (A) To introduce into the human body any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (B) To enhance the effect on the human body of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (C) To conceal any quantity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; or (D) To test the strength, effectiveness, or purity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state. (4) "Knowingly" means having general knowledge that a machine, instrument, tool, item of equipment, contrivance, or device is a drug related object or having reasonable grounds to believe that any such object is or may, to an average person, appear to be a drug related object. -
Guidance for the Format and Content of the Protocol of Non-Interventional
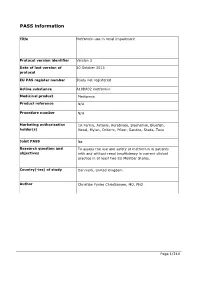
PASS information Title Metformin use in renal impairment Protocol version identifier Version 2 Date of last version of 30 October 2013 protocol EU PAS register number Study not registered Active substance A10BA02 metformin Medicinal product Metformin Product reference N/A Procedure number N/A Marketing authorisation 1A Farma, Actavis, Aurobindo, Biochemie, Bluefish, holder(s) Hexal, Mylan, Orifarm, Pfizer, Sandoz, Stada, Teva Joint PASS No Research question and To assess the use and safety of metformin in patients objectives with and without renal insufficiency in current clinical practice in at least two EU Member States. Country(-ies) of study Denmark, United Kingdom Author Christian Fynbo Christiansen, MD, PhD Page 1/214 Marketing authorisation holder(s) Marketing authorisation N/A holder(s) MAH contact person N/A Page 2/214 1. Table of Contents PASS information .......................................................................................................... 1 Marketing authorisation holder(s) .................................................................................... 2 1. Table of Contents ...................................................................................................... 3 2. List of abbreviations ................................................................................................... 4 3. Responsible parties .................................................................................................... 5 4. Abstract .................................................................................................................. -

Levobetaxolol Hydrochloride
1882 Miotics Mydriatics and Antiglaucoma Drugs if necessary 5 to 10 minutes later. In the treatment of uveitis Herpes simplex dendritic keratitis developed in 2 patients during Levobetaxolol Hydrochloride (USAN, rINNM) ⊗ (p.1515), the eye drops should be instilled two or three times dai- latanoprost therapy.3 The author suggested that the biochemical ly, or up to every 3 to 4 hours if required. changes in the cornea caused by latanoprost may predispose to AL-1577A (levobetaxolol or levobetaxolol hydrochloride); Hid- herpes keratitis. rocloruro de levobetaxolol; Lévobétaxolol, Chlorhydrate de; The BNFC recommends that eye drops containing 0.5% homat- Levobetaxololi Hydrochloridum. (−)-(S)-1-{p-[2-(Cyclopropyl- ropine hydrobromide are used once daily or on alternate days for 1. Wardrop DRA, Wishart PK. Latanoprost and cystoid macular methoxy)ethyl]phenoxy}-3-isopropylaminopropan-2-ol hydro- uveitis in children aged 3 months to 2 years; older children may oedema in a pseudophake. Br J Ophthalmol 1998; 82: 843–4. be given 1 or 2% eye drops twice daily. 2. Stewart O, et al. Bilateral optic disc oedema associated with chloride. latanoprost. Br J Ophthalmol 1999; 83: 1092–3. Левобетаксолола Гидрохлорид Homatropine has also been used as the quaternary ammonium 3. Ekatomatis P. Herpes simplex dendritic keratitis after treatment methobromide derivative in the treatment of gastrointestinal with latanoprost for primary open angle glaucoma. Br J Ophthal- C18H29NO3,HCl = 343.9. spasm and as an adjunct in peptic ulcer disease; homatropine mol 2001; 85: 1008–9. CAS — 93221-48-8 (levobetaxolol); 116209-55-3 (levo- betaxolol hydrochloride). methobromide has also been included in preparations used for Systemic effects. -

Canine Red Eye Elizabeth Barfield Laminack, DVM; Kathern Myrna, DVM, MS; and Phillip Anthony Moore, DVM, Diplomate ACVO
PEER REVIEWED Clinical Approach to the CANINE RED EYE Elizabeth Barfield Laminack, DVM; Kathern Myrna, DVM, MS; and Phillip Anthony Moore, DVM, Diplomate ACVO he acute red eye is a common clinical challenge for tion of the deep episcleral vessels, and is characterized general practitioners. Redness is the hallmark of by straight and immobile episcleral vessels, which run Tocular inflammation; it is a nonspecific sign related 90° to the limbus. Episcleral injection is an external to a number of underlying diseases and degree of redness sign of intraocular disease, such as anterior uveitis and may not reflect the severity of the ocular problem. glaucoma (Figures 3 and 4). Occasionally, episcleral Proper evaluation of the red eye depends on effective injection may occur in diseases of the sclera, such as and efficient diagnosis of the underlying ocular disease in episcleritis or scleritis.1 order to save the eye’s vision and the eye itself.1,2 • Corneal Neovascularization » Superficial: Long, branching corneal vessels; may be SOURCE OF REDNESS seen with superficial ulcerative (Figure 5) or nonul- The conjunctiva has small, fine, tortuous and movable vessels cerative keratitis (Figure 6) that help distinguish conjunctival inflammation from deeper » Focal deep: Straight, nonbranching corneal vessels; inflammation (see Ocular Redness algorithm, page 16). indicates a deep corneal keratitis • Conjunctival hyperemia presents with redness and » 360° deep: Corneal vessels in a 360° pattern around congestion of the conjunctival blood vessels, making the limbus; should arouse concern that glaucoma or them appear more prominent, and is associated with uveitis (Figure 4) is present1,2 extraocular disease, such as conjunctivitis (Figure 1). -

Ophthalmic Agents, Intraocular Pressure (IOP)-Modifying
Therapeutic Class Overview Ophthalmic Agents, Intraocular Pressure (IOP)-Modifying INTRODUCTION Glaucoma is an optic neuropathy that causes gradual degeneration of the cells making up the optic nerve. Glaucoma is among the leading causes of blindness worldwide, and in 2020, an estimated 3.2 million people worldwide are anticipated to be blind due to glaucoma (Flaxman et al 2017). Open-angle glaucoma is the most common form; other forms include angle-closure, congenital, and secondary glaucoma (Jacobs 2018[a]). Patients with open-angle glaucoma initially experience peripheral visual field loss, followed by central field loss, which may progress to irreversible blindness if untreated (Jacobs 2018[a]). The exact etiology of open-angle glaucoma is unknown (Jacobs 2018[a]). Major risk factors for developing open-angle glaucoma include advanced age, African or Hispanic/Latino descent, elevated intraocular pressure (IOP), family history of glaucoma, low ocular perfusion pressure, type 2 diabetes mellitus, and myopia (Ellis et al 2000, Girkin et al 2004, Lesk et al 2007, Prum et al 2016). Elevated IOP is the only major risk factor for glaucoma that is treatable. Available evidence suggests that lowering IOP inhibits or reduces the progression of optic nerve damage (Jacobs 2018[a]). Treatment may be initiated in patients with a raised IOP despite having no visual field loss or optic nerve damage (Jacobs 2018[a]). An IOP > 22 to 25 mmHg is generally considered to be elevated and would be treated by most clinicians; however, this number varies according to screening methods, risk factors, and disease progression (Jacobs 2018[b]). The target IOP should be individualized based on response to therapy and disease progression in order to maintain IOP within a range that is unlikely to adversely affect patients’ health-related quality of life (Jacobs 2018[b]). -

Antiglaucoma Compositions Containing Combinations of Alpha-2 Agonists and Beta-Blockers
~" ' MM II II II MM II II II Ml II II I II J European Patent Office _ _ _ © Publication number: 0 365 662 B1 Office europeen* des.. brevets , © EUROPEAN PATENT SPECIFICATION © Date of publication of patent specification: 09.03.94 © Int. CI.5: A61 K 31/47 © Application number: 89905874.7 @ Date of filing: 26.04.89 © International application number: PCT/US89/01994 © International publication number: WO 89/10126 (02.11.89 89/26) (54) ANTIGLAUCOMA COMPOSITIONS CONTAINING COMBINATIONS OF ALPHA-2 AGONISTS AND BETA-BLOCKERS. ® Priority: 26.04.88 US 186504 1075-1078 @ Date of publication of application: J. of Cardiovascular Pharmacology, vol. 2, 02.05.90 Bulletin 90/18 suppl. 1, 1980, S. 21-28 © Publication of the grant of the patent: © Proprietor: ALCON LABORATORIES INC 09.03.94 Bulletin 94/10 6201 South Freeway Ft. Worth, TX 76134(US) © Designated Contracting States: AT BE CH DE FR GB IT LI LU NL SE @ Inventor: DE SANTIS, Louis, M. 2316 Wlnton Terrace West © References cited: Fort Worth, TX 761 09(US) US-A- 4 455 317 US-A- 4 515 800 © Representative: Jump, Timothy John Simon et AM A DRUG EVALUATION, 2nd ed., 1973; al "Agent used to treat Glaucoma", pp. 675-686. Venner Shipley & Co. 20 Little Britain GLAUCOMA, vol. 1, no. 1, February 1979; London EC1A 7DH (GB) 00 S.SUGAR, pp. 9-15. CM CO Ophthalmology, vol. 96, no. 1, 1989, p. 3-7 CO m Ophthalmology, vol. 98, no. 7, 1991, p. CO 00 Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. -

Glaucoma Medical Treatment: Philosophy, Principles and Practice
Glaucoma medical CLIVE MIGDAL treatment: philosophy, principles and practice Abstract assessment of these parameters. Indeed There have been numerous recent advances in compounds are under evaluation that affect the the management of glaucoma, not least the function of the optic nerve (via improved blood development of new drugs to help manage supply or improved neuronal cell physiology) raised intraocular pressure. In addition, the but may or may not lower lOP. It may even be concepts of improving blood flow to the optic possible in the future to therapeutically alter the nerve head and neuroprotection are currently human genome, genetically deliver provoking considerable interest. This article neuroprotective substances or aid regeneration considers the aims and philosophy of of the optic nerve axons. glaucoma drug therapy, summarises some of The main aim of glaucoma therapy must still the basic facts and principles of modem be the preservation of visual function. At the glaucoma medications, and suggests a same time, the therapy should not have adverse practical approach to the choice of therapy. side effects and should not affect the quality of life of the patient (by causing either side effects Key words Blood flow, Intraocular pressure, or inconvenience and disruption of daily Neuroprotection, Primary open angle glaucoma, Topical medications lifestyle). The cost of the therapy, both direct and indirect, must also be taken into consideration.s Currently, typical glaucoma management Philosophy consists of lowering the lOP to a satisfactory Primary open-angle glaucoma is a complex and safe target leve1.6 To determine the success disease for which a number of risk factors have of this treatment, the patient must be followed been identified, including intraocular pressure, long-term with routine assessment of lOP, discs age, race and family history.l,2 Due to our and fields to exclude progressive damage. -

Betaxolol Ophthalmic Solution As Alternative Treatment for Patients with Timolol Allergy: a Case Report
Case Report Betaxolol Ophthalmic Solution as Alternative Treatment for Patients with Timolol Allergy: A Case Report Olivia Müllertz 1,*, Jette Jacobsen 2, Jacob P. Thyssen 3, Anna Horwitz 1,4 and Miriam Kolko 1,4,* 1 Department of Drug Design and Pharmacology, University of Copenhagen, 2100 Copenhagen, Denmark; [email protected] 2 Department of Pharmacy, University of Copenhagen, 2100 Copenhagen, Denmark; [email protected] 3 Department of Dermatology and Allergy, Herlev-Gentofte Hospital, 2900 Hellerup, Denmark; [email protected] 4 Department of Ophthalmology, Copenhagen University Hospital, Rigshospitalet, 2600 Glostrup, Denmark * Correspondence: [email protected] (O.M.); [email protected] (M.K.) Received: 24 March 2020; Accepted: 25 June 2020; Published: 30 July 2020 Abstract: Background: To establish if an allergy towards all β-blockers, as a group, should be assumed, if an allergic reaction is observed while using one specific β-blocking agent. Case presentation: The non-selective β-blocker timolol caused a severe allergic ocular reaction in a non-atopic patient with advanced primary open-angle glaucoma. Results: A patch test confirmed timolol allergy. No allergic reaction to other anti-glaucomatous topical drugs was observed, and treatment with the selective β-blocker betaxolol was successfully initiated. Conclusion: Allergy to the non-selective β-blocker timolol does not necessarily predict allergy to the selective β-blocker betaxolol, and betaxolol should therefore not be excluded as an alternative treatment. Keywords: betaxolol; timolol; β-blocker; allergy 1. Introduction Drug allergies are a class of unpredictable adverse reactions that are not related to the pharmacological effect of a drug, and can occur at subtherapeutic doses. -

Download Product Insert (PDF)
PRODUCT INFORMATION Levobetaxolol (hydrochloride) Item No. 33435 CAS Registry No.: 116209-55-3 Formal Name: (2S)-1-[4-[2-(cyclopropylmethoxy)ethyl] phenoxy]-3-[(1-methylethyl)amino]-2- propanol, monohydrochloride O Synonym: (S)-Betaxolol MF: C18H29NO3 • HCl • HCl FW: 343.9 N O Purity: ≥98% H OH Supplied as: A crystalline solid Storage: -20°C Stability: ≥2 years Information represents the product specifications. Batch specific analytical results are provided on each certificate of analysis. Laboratory Procedures Levobetaxolol (hydrochloride) is supplied as a crystalline solid. A stock solution may be made by dissolving the levobetaxolol (hydrochloride) in the solvent of choice, which should be purged with an inert gas. Levobetaxolol (hydrochloride) is soluble in organic solvents such as ethanol, DMSO, and dimethyl formamide (DMF). The solubility of levobetaxolol (hydrochloride) in ethanol is approximately 1 mg/ml and approximately 10 mg/ml in DMSO and DMF. Further dilutions of the stock solution into aqueous buffers or isotonic saline should be made prior to performing biological experiments. Ensure that the residual amount of organic solvent is insignificant, since organic solvents may have physiological effects at low concentrations. Organic solvent-free aqueous solutions of levobetaxolol (hydrochloride) can be prepared by directly dissolving the crystalline solid in aqueous buffers. The solubility of levobetaxolol (hydrochloride) in PBS (pH 7.2) is approximately 10 mg/ml. We do not recommend storing the aqueous solution for more than one day. Description Levobetaxolol is an isomer of betaxolol (Item No. 18625) and antagonist of the β1-adrenergic receptor 1 (β1-AR; Ki = 0.76 nM for the human receptor). It is selective for β1-ARs over β2-ARs (Ki = 32.6 nM), as well as a panel of 89 additional receptors (IC50s = >1 µM).