Brimonidine Tartrate; Brinzolamide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Intraocular Pressure Rise After Phacoemulsification Prophylactic
British Journal of Ophthalmology 1995; 79: 809-813 809 Intraocular pressure rise after phacoemulsification Br J Ophthalmol: first published as 10.1136/bjo.79.9.809 on 1 September 1995. Downloaded from with posterior chamber lens implantation: effect of prophylactic medication, wound closure, and surgeon's experience Thomas G Bomer, Wolf-Dietrich A Lagreze, Jens Funk Abstract pressure rises usually occur between 6 and 8 Aims-A prospective clinical trial was hours after surgery.6 carried out to evaluate the effect of Various antiglaucomatous agents have been prophylactic medication, the technique of used to prevent the intraocular pressure rise wound closure, and the surgeon's experi- after cataract extraction. Oral acetazolamide15 16 ence on the intraocular pressure rise after and topical timolol'6-19 lowered the pressure cataract extraction. rise in the early period after intracapsular and Methods-In 100 eyes, the intraocular extracapsular cataract extraction. Levobunolol pressure was measured before as well as proved to be superior to timolol 4-7 hours 2-4, 5-7, and 22-24 hours after phaco- after extracapsular cataract extraction.20 Apra- emulsification and posterior chamber lens clonidine lowered the intraocular pressure rise implantation. Each of 25 patients received after uncomplicated phacoemulsification21 and either 1% topical apraclonidine, 0.5%/o extracapsular cataract extraction22 23 when topical levobunolol, 500 mg oral acetazo- given 30 minutes to 1 hour before surgery, lamide, or placebo. Forty four eyes were whereas immediate postoperative treatment operated with sclerocorneal sutureless with apraclonidine was ineffective.22 24 Miotics tunnel and 56 eyes with corneoscleral are frequently used to promote miosis and incision and suture. -

Table 1. Glaucoma Medications: Mechanisms, Dosing and Precautions Brand Generic Mechanism of Action Dosage/Avg
OPTOMETRIC STUDY CENTER Table 1. Glaucoma Medications: Mechanisms, Dosing and Precautions Brand Generic Mechanism of Action Dosage/Avg. % Product Sizes Side Effects Warnings Reduction CHOLINERGIC AGENTS Direct Pilocarpine (generic) Pilocarpine 1%, 2%, 4% Increases trabecular outflow BID-QID/15-25% 15ml Headache, blurred vision, myopia, retinal detachment, bronchiole constriction, Angle closure, shortness of breath, retinal narrowing of angle detachment Indirect Phospholine Iodide (Pfizer) Echothiophate iodide 0.125% Increases trabecular outflow QD-BID/15-25% 5ml Same as above plus cataractogenic iris cysts in children, pupillary block, Same as above, plus avoid prior to any increased paralysis with succinylcholine general anesthetic procedure ALPHA-2 AGONISTS Alphagan P (Allergan) Brimonidine tartrate 0.1%, 0.15% with Purite Decreases aqueous production, increases BID-TID/up to 26% 5ml, 10ml, 15ml Dry mouth, hypotension, bradycardia, follicular conjunctivitis, ocular irritation, Monitor for shortness of breath, dizziness, preservative uveoscleral outflow pruritus, dermatitis, conjunctival blanching, eyelid retraction, mydriasis, drug ocular redness and itching, fatigue allergy Brimonidine tartrate Brimonidine tartrate 0.15%, 0.2% Same as above Same as above 5ml, 10ml Same as above Same as above (generic) Iopidine (Novartis) Apraclonidine 0.5% Decreases aqueous production BID-TID/up to 25% 5ml, 10ml Same as above but higher drug allergy (40%) Same as above BETA-BLOCKERS Non-selective Betagan (Allergan) Levobunolol 0.25%, 0.5% Decreases -

AZARGA, INN-Brinzolamide/Timolol
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 1. NAME OF THE MEDICINAL PRODUCT AZARGA 10 mg/ml + 5 mg/ml eye drops, suspension 2. QUALITATIVE AND QUANTITATIVE COMPOSITION One ml of suspension contains 10 mg brinzolamide and 5 mg timolol (as timolol maleate). Excipient with known effect: One ml of suspension contains 0.10 mg benzalkonium chloride. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Eye drops, suspension (eye drops) White to off-white uniform suspension, pH 7.2 (approximately). 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Decrease of intraocular pressure (IOP) in adult patients with open-angle glaucoma or ocular hypertension for whom monotherapy provides insufficient IOP reduction (see section 5.1). 4.2 Posology and method of administration Posology Use in adults, including the elderly The dose is one drop of AZARGA in the conjunctival sac of the affected eye(s) twice daily. When using nasolacrimal occlusion or closing the eyelids, the systemic absorption is reduced. This may result in a decrease in systemic side effects and an increase in local activity (see section 4.4). If a dose is missed, treatment should be continued with the next dose as planned. The dose should not exceed one drop in the affected eye (s) twice daily. When substituting another ophthalmic antiglaucoma medicinal product with AZARGA, the other medicinal product should be discontinued and AZARGA should be started the following day. Special populations Paediatric population The safety and efficacy of AZARGA in children and adolescents aged 0 to 18 years have not yet been established. -

Title 16. Crimes and Offenses Chapter 13. Controlled Substances Article 1
TITLE 16. CRIMES AND OFFENSES CHAPTER 13. CONTROLLED SUBSTANCES ARTICLE 1. GENERAL PROVISIONS § 16-13-1. Drug related objects (a) As used in this Code section, the term: (1) "Controlled substance" shall have the same meaning as defined in Article 2 of this chapter, relating to controlled substances. For the purposes of this Code section, the term "controlled substance" shall include marijuana as defined by paragraph (16) of Code Section 16-13-21. (2) "Dangerous drug" shall have the same meaning as defined in Article 3 of this chapter, relating to dangerous drugs. (3) "Drug related object" means any machine, instrument, tool, equipment, contrivance, or device which an average person would reasonably conclude is intended to be used for one or more of the following purposes: (A) To introduce into the human body any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (B) To enhance the effect on the human body of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (C) To conceal any quantity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; or (D) To test the strength, effectiveness, or purity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state. (4) "Knowingly" means having general knowledge that a machine, instrument, tool, item of equipment, contrivance, or device is a drug related object or having reasonable grounds to believe that any such object is or may, to an average person, appear to be a drug related object. -

NEW ZEALAND DATA SHEET 1. PRODUCT NAME IOPIDINE® (Apraclonidine Hydrochloride) Eye Drops 0.5%
NEW ZEALAND DATA SHEET 1. PRODUCT NAME IOPIDINE® (apraclonidine hydrochloride) Eye Drops 0.5%. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each mL of Iopidine Eye Drops 0.5% contains apraclonidine hydrochloride 5.75 mg, equivalent to apraclonidine base 5 mg. Excipient with known effect Benzalkonium chloride 0.1 mg per 1 mL as a preservative. For the full list of excipients, see section 6.1. 2. PHARMACEUTICAL FORM Eye drops, solution, sterile, isotonic. 4. CLINICAL PARTICULARS 4.1. Therapeutic indications Iopidine Eye Drops 0.5% are indicated for short-term adjunctive therapy in patients on maximally tolerated medical therapy who require additional IOP reduction. Patients on maximally tolerated medical therapy who are treated with Iopidine Eye Drops 0.5% to delay surgery should have frequent follow up examinations and treatment should be discontinued if the intraocular pressure rises significantly. The addition of Iopidine Eye Drops 0.5% to patients already using two aqueous suppressing drugs (i.e. beta-blocker plus carbonic anhydrase inhibitor) as part of their maximally tolerated medical therapy may not provide additional benefit. This is because apraclonidine is an aqueous-suppressing drug and the addition of a third aqueous suppressant may not significantly reduce IOP. The IOP-lowering efficacy of Iopidine Eye Drops 0.5% diminishes over time in some patients. This loss of effect, or tachyphylaxis, appears to be an individual occurrence with a variable time of onset and should be closely monitored. The benefit for most patients is less than one month. 4.2. Dose and method of administration Dose One drop of Iopidine Eye Drops 0.5% should be instilled into the affected eye(s) three times per day. -

Levobetaxolol Hydrochloride
1882 Miotics Mydriatics and Antiglaucoma Drugs if necessary 5 to 10 minutes later. In the treatment of uveitis Herpes simplex dendritic keratitis developed in 2 patients during Levobetaxolol Hydrochloride (USAN, rINNM) ⊗ (p.1515), the eye drops should be instilled two or three times dai- latanoprost therapy.3 The author suggested that the biochemical ly, or up to every 3 to 4 hours if required. changes in the cornea caused by latanoprost may predispose to AL-1577A (levobetaxolol or levobetaxolol hydrochloride); Hid- herpes keratitis. rocloruro de levobetaxolol; Lévobétaxolol, Chlorhydrate de; The BNFC recommends that eye drops containing 0.5% homat- Levobetaxololi Hydrochloridum. (−)-(S)-1-{p-[2-(Cyclopropyl- ropine hydrobromide are used once daily or on alternate days for 1. Wardrop DRA, Wishart PK. Latanoprost and cystoid macular methoxy)ethyl]phenoxy}-3-isopropylaminopropan-2-ol hydro- uveitis in children aged 3 months to 2 years; older children may oedema in a pseudophake. Br J Ophthalmol 1998; 82: 843–4. be given 1 or 2% eye drops twice daily. 2. Stewart O, et al. Bilateral optic disc oedema associated with chloride. latanoprost. Br J Ophthalmol 1999; 83: 1092–3. Левобетаксолола Гидрохлорид Homatropine has also been used as the quaternary ammonium 3. Ekatomatis P. Herpes simplex dendritic keratitis after treatment methobromide derivative in the treatment of gastrointestinal with latanoprost for primary open angle glaucoma. Br J Ophthal- C18H29NO3,HCl = 343.9. spasm and as an adjunct in peptic ulcer disease; homatropine mol 2001; 85: 1008–9. CAS — 93221-48-8 (levobetaxolol); 116209-55-3 (levo- betaxolol hydrochloride). methobromide has also been included in preparations used for Systemic effects. -

ALPHAGAN® 0.2% Safely and Effectively
NDA 20613/S-031 Page 4 HIGHLIGHTS OF PRESCRIBING INFORMATION ___________ ___________ ADVERSE REACTIONS Most common adverse reactions occurring in These highlights do not include all the information needed approximately 10 to 30% of patients receiving brimonidine to use ALPHAGAN® 0.2% safely and effectively. See full ophthalmic solution 0.2% included oral dryness, ocular prescribing information for ALPHAGAN® 0.2%. hyperemia, burning and stinging, headache, blurring, foreign body sensation, fatigue/drowsiness, conjunctival ALPHAGAN® (brimonidine tartrate ophthalmic solution) follicles, ocular allergic reactions, and ocular pruritus. (6.1) 0.2% For Topical Ophthalmic Use To report SUSPECTED ADVERSE REACTIONS, Initial U.S. Approval: 1996 contact Allergan at 1-800-433-8871 or the FDA at 1 800-FDA-1088 or www.fda.gov/medwatch. _________ _________ INDICATIONS AND USAGE ___________ ____________ DRUG INTERACTIONS ALPHAGAN® 0.2% is an alpha adrenergic agonist indicated Antihypertensives/cardiac glycosides may lower blood for lowering intraocular pressure (IOP) in patients with open- pressure. (7.1) angle glaucoma or ocular hypertension. (1) ________ ________ Use with CNS depressants may result in an additive or DOSAGE AND ADMINISTRATION potentiating effect. (7.2) One drop in the affected eye(s), three times daily, Tricyclic antidepressants may potentially blunt the approximately 8 hours apart. (2) hypotensive effect of systemic clonidine. (7.3) _______ _______ DOSAGE FORMS AND STRENGTHS Monoamine oxidase inhibitors may result in increased Solution containing 2 mg/mL brimonidine tartrate. (3) hypotension. (7.4) _________ _____________ _______ ______ CONTRAINDICATIONS USE IN SPECIFIC POPULATIONS Neonates and infants (under the age of 2 years). (4.1) Use with caution in children > 2 years of age. -

New Medicines Committee Briefing March 2015 Simbrinza
New Medicines Committee Briefing March 2015 Simbrinza® (Brinzolamide 10mg + Brimonidine tartrate 2mg/ml) for treatment of elevated intraocular pressure (IOP) in adult patients with open-angle glaucoma or ocular hypertension. Simbrinza® is to be reviewed for use within: Primary Care Secondary Care Summary: Simbrinza® is a combination of brinzolamide which is a carbonic anhydrase (CA-II) inhibitor and brimonidine which is an alpha-2 adrenergic agonist. Simbrinza® is licensed for the treatment of elevated IOP in adult patients with open-angle glaucoma or ocular hypertension whom failed on mono-therapy. Simbrinza® is administered twice daily in the affected eye(s). Simbrinza® is the first combination therapy which does not contain a beta-blocker. SMC has accepted Simbrinza® for use within NHS Scotland, as there is no significant additional cost associated with the combination product compared with its individual components. One trial indicated that fixed combination of Simbrinza® administered BD had a significantly greater IOP lowering effect compared to either brinzolamide 1% or brimonidine 0.2% alone and displayed a safety profile consistent with its individual components. Another trial noted Simbrinza® to be non-inferior to separate administration of brimonidine and brinzolamide less than 10 min apart to prevent wash out. 1 Formulary application: Opthamology department: Consultant submitting application: Mr Lynval Jones (Consultant Ophthalmologist, Glaucoma) Clinical Director supporting application: Gareth Rowland (Clinical Director of Specialised Surgery) Mr Jones has requested for Simbrinza® to be considered for inclusion in the North Staffordshire Joint Formulary for the treatment of elevated intraocular pressure (IOP) in adult patients with open-angle glaucoma or ocular hypertension who have not responded to monotherapy. -

View Board, Austin, Texas, As Well As the Ethics Com- Pendent Examiner in the Same Room with the Same Instru- Mittee of Eye Center and Research, Aseer, Saudi Arabia
Abdelkader and Kaufman Eye and Vision (2016) 3:31 DOI 10.1186/s40662-016-0065-3 RESEARCH Open Access Clinical outcomes of combined versus separate carbachol and brimonidine drops in correcting presbyopia Almamoun Abdelkader1* and Herbert E. Kaufman2 Abstract Background: To test and compare in a masked fashion the efficacy of using a parasympathomimetic drug (3% carbachol) and an alpha-2 agonist (0.2% brimonidine) in both combined and separate forms to create optically beneficial miosis to pharmacologically improve vision in presbyopia. Methods: A prospective, double-masked, randomized, controlled clinical trial was conducted. Ten naturally emmetropic and presbyopic subjects between 42 and 58 years old with uncorrected distance visual acuity of at least 20/20 in both eyes without additional ocular pathology were eligible for inclusion. All subjects received 3% carbachol and 0.2% brimonidine in both combined and separate forms, 3% carbachol alone and 0.2% brimonidine (control) alone in their non-dominant eye in a crossover manner with one week washout between tests. The subjects’ pupil sizes and both near and distance visual acuities will be evaluated pre- and post-treatment at 1, 2, 4, and 8 h, by a masked examiner at the same room illumination. Results: Statistically significant improvement in mean near visual acuity (NVA) was achieved in all subjects who received combined 3% carbachol and 0.2% brimonidine in the same formula compared with those who received separate forms or carbachol alone or brimonidine alone (P < 0.0001). Conclusion: Based on the data, the combined solution demonstrated greater efficacy than the other solutions that were tested. -

WO 2014/066775 Al 1 May 2014 (01.05.2014) W P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2014/066775 Al 1 May 2014 (01.05.2014) W P O PCT (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61F 9/00 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/US20 13/066834 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 25 October 2013 (25.10.201 3) KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (25) Filing Language: English OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (26) Publication Language: English SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, (30) Priority Data: ZW. 61/719,144 26 October 2012 (26. 10.2012) US (84) Designated States (unless otherwise indicated, for every (71) Applicant: FORSIGHT VISION5, INC. [US/US]; 191 kind of regional protection available): ARIPO (BW, GH, Jefferson Drive, Menlo Park, CA 94025 (US). GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, (72) Inventors: RUBIN, Anne, Brody; 191 Jefferson Drive, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, Menlo Park, CA 94025 (US). -
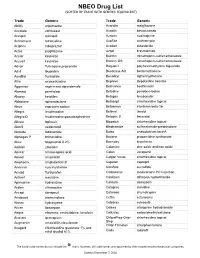
Drug List (SORTED by TRADE with GENERIC EQUIVALENT)
NBEO Drug List (SORTED BY TRADE WITH GENERIC EQUIVALENT) Trade Generic Trade Generic Abilify aripiprazole Avandia rosiglitazone Accolate zafirlukast Avastin bevacizumab Accupril quinapril Azasan azathioprine Achromycin tetracycline AzaSite azithromycin Aciphex rabeprazole Avodart dutasteride Actos pioglitazone Azopt brinzolamide Acular ketorolac Bactrim trimethoprim-sulfamethoxazole Acuvail ketorolac Bactrim DS trimethoprim-sulfamethoxazole Advair fluticasone propionate Baquacil polyhexamethylene biguanide Advil ibuprofen Beconase AQ beclomethasone AeroBid flunisolide Benadryl diphenhydramine Afrin oxymetazoline Bepreve Bepotastine besilate Aggrenox aspirin and dipyridamole Besivance besifloxacin Alamast pemirolast Betadine povidone-iodine Alaway ketotifen Betagan levobunolol Aldactone spironolactone Betasept chlorhexidine topical Aleve naproxen sodium Betaseron interferon beta-1b Allegra fexofenadine Betimol timolol Allegra-D fexofenadine-pseudoephedrine Betoptic S betaxolol Alluvia lopinavir Biopatch chlorhexidine topical Alocril nedocromil Blephamide sulfacetamide-prednisolone Alomide lodoxamide Botox onabotulinum toxinA Alphagan P brimonidine Brolene propamidine isethionate Alrex loteprednol 0.2% Bromday bromfenac Ambien zolpidem Calamine zinc oxide and iron oxide Amicar aminocaproic acid Calan verapamil Amoxil amoxicillin Calgon Vesta chlorhexidine topical Amphocin amphotericin B Capoten captopril Anectine succinylcholine Carafate sucralfate Ansaid flurbiprofen Carbocaine mepivacaine HCl injection Antivert meclizine Cardizem diltiazem -

IOPIDINE® 0.5% (Apraclonidine Ophthalmic Solution) 0.5% As Base
IOPIDINE® 0.5% (apraclonidine ophthalmic solution) 0.5% As Base DESCRIPTION: IOPIDINE® 0.5% Ophthalmic Solution contains apraclonidine hydrochloride, an alpha adrenergic agonist, in a sterile isotonic solution for topical application to the eye. Apraclonidine hydrochloride is a white to off-white powder and is highly soluble in water. Its chemical name is 2-[(4-amino-2,6 dichlorophenyl) imino]imidazolidine monohydrochloride with an empirical formula of C9H11Cl3N4 and a molecular weight of 281.57. The chemical structure of apraclonidine hydrochloride is: Each mL of IOPIDINE 0.5% Ophthalmic Solution contains: Active: apraclonidine hydrochloride 5.75 mg equivalent to apraclonidine base 5 mg; . Inactives: sodium chloride, sodium acetate, sodium hydroxide and/or hydrochloric acid (pH 4.4-7.8), purified water and benzalkonium chloride 0.01% (preservative) CLINICAL PHARMACOLOGY: Apraclonidine hydrochloride is a relatively selective alpha- 2-adrenergic agonist. When instilled in the eye, IOPIDINE 0.5% Ophthalmic Solution, has the action of reducing elevated, as well as normal, intraocular pressure (IOP), whether or not accompanied by glaucoma. Ophthalmic apraclonidine has minimal effect on cardiovascular parameters. Elevated IOP presents a major risk factor in glaucomatous field loss. The higher the level of IOP, the greater the likelihood of optic nerve damage and visual field loss. IOPIDINE 0.5% Ophthalmic Solution has the action of reducing IOP. The onset of action of apraclonidine can usually be noted within one hour, and maximum IOP reduction occurs about three hours after instillation. Aqueous fluorophotometry studies demonstrate that apraclonidine's predominant mechanism of action is reduction of aqueous flow via stimulation of the alpha-adrenergic system.