The Role of ACE2 in COVID-19'S Tissue Damaging Effects
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mobile Genetic Elements in Streptococci
Curr. Issues Mol. Biol. (2019) 32: 123-166. DOI: https://dx.doi.org/10.21775/cimb.032.123 Mobile Genetic Elements in Streptococci Miao Lu#, Tao Gong#, Anqi Zhang, Boyu Tang, Jiamin Chen, Zhong Zhang, Yuqing Li*, Xuedong Zhou* State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, PR China. #Miao Lu and Tao Gong contributed equally to this work. *Address correspondence to: [email protected], [email protected] Abstract Streptococci are a group of Gram-positive bacteria belonging to the family Streptococcaceae, which are responsible of multiple diseases. Some of these species can cause invasive infection that may result in life-threatening illness. Moreover, antibiotic-resistant bacteria are considerably increasing, thus imposing a global consideration. One of the main causes of this resistance is the horizontal gene transfer (HGT), associated to gene transfer agents including transposons, integrons, plasmids and bacteriophages. These agents, which are called mobile genetic elements (MGEs), encode proteins able to mediate DNA movements. This review briefly describes MGEs in streptococci, focusing on their structure and properties related to HGT and antibiotic resistance. caister.com/cimb 123 Curr. Issues Mol. Biol. (2019) Vol. 32 Mobile Genetic Elements Lu et al Introduction Streptococci are a group of Gram-positive bacteria widely distributed across human and animals. Unlike the Staphylococcus species, streptococci are catalase negative and are subclassified into the three subspecies alpha, beta and gamma according to the partial, complete or absent hemolysis induced, respectively. The beta hemolytic streptococci species are further classified by the cell wall carbohydrate composition (Lancefield, 1933) and according to human diseases in Lancefield groups A, B, C and G. -

Gutless Adenovirus: Last-Generation Adenovirus for Gene Therapy
Gene Therapy (2005) 12, S18–S27 & 2005 Nature Publishing Group All rights reserved 0969-7128/05 $30.00 www.nature.com/gt CONFERENCE PAPER Gutless adenovirus: last-generation adenovirus for gene therapy R Alba1, A Bosch1 and M Chillon1,2 1Gene Therapy Laboratory, Department of Biochemistry and Molecular Biology, Center of Animal Biotechnology and Gene Therapy (CBATEG), Universitat Auto`noma de Barcelona, Bellaterra, Spain; and 2Institut Catala` de Recerca i Estudis Avanc¸ats (ICREA), Barcelona, Spain Last-generation adenovirus vectors, also called helper-depen- viral coding regions, gutless vectors require viral proteins dent or gutless adenovirus, are very attractive for gene therapy supplied in trans by a helper virus. To remove contamination because the associated in vivo immune response is highly by a helper virus from the final preparation, different systems reduced compared to first- and second-generation adenovirus based on the excision of the helper-packaging signal have vectors, while maintaining high transduction efficiency and been generated. Among them, Cre-loxP system is mostly tropism. Nowadays, gutless adenovirus is administered in used, although contamination levels still are 0.1–1% too high different organs, such as the liver, muscle or the central to be used in clinical trials. Recently developed strategies to nervous system achieving high-level and long-term transgene avoid/reduce helper contamination were reviewed. expression in rodents and primates. However, as devoid of all Gene Therapy (2005) 12, S18–S27. doi:10.1038/sj.gt.3302612 Keywords: adenovirus; gutless; helper-dependent vectors; in vivo gene therapy Introduction clinical for more information). Nowadays, adenovirus vectors are applied to treat cancer, monogenic disorders, Gene therapy for most genetic diseases requires expres- vascular diseases and others complications. -

Viral Vectors and Biological Safety
Viral Vectors and Biological Safety Viral vectors are often designed so that they can enter human cells and deliver genes of interest. Viral vectors are usually replication-deficient – genes necessary for replication of the virus are removed from the vector and supplied separately through plasmids, helper virus, or packaging cell lines. There are several biosafety concerns that arise with the use of viral vectors including: 1) Tropism (host range) – viral vectors that can enter (infect) human cells are often used. 2) Replication-deficient viral vectors can gain back the deleted genes required for replication (become replication-competent) through recombination – referred to as replication-competent virus (RCV) breakthroughs. 3) Genes may be expressed in tissues and/or organisms where they are normally not expressed. In the case of some genes such as oncogenes, this could have far-reaching negative consequences. When evaluating safety for use of viral vectors, a number of factors need to be considered including: Risk Group (RG) of the organism; tropism (organism and tissue); route of transmission; whether the virus integrates into the host genome; and the specific gene(s) being introduced. Please contact the Office of Biological Safety (OBS) for more information on physical barriers and safety practices to use with specific viral vectors. This article concentrates on biological barriers that can be employed to improve safety when using viral vectors. Viral vectors frequently used are: • Retrovirus/lentivirus • Adenovirus • Adeno-associated virus (AAV) • Poxvirus • Herpes virus • Alphavirus • Baculovirus Amphotropic murine leukemia virus (MLV) – also called Moloney murine leukemia virus (MMLV) – and adenovirus are common viral vectors used to introduce genes into human cells. -
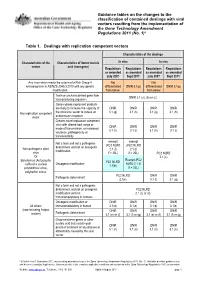
Guidance Tables on the Changes To
Guidance tables on the changes to the classification of contained dealings with viral vectors resulting from the implementation of the Gene Technology Amendment Regulations 2011 (No. 1)* Table 1. Dealings with replication competent vectors Characteristics of the dealings Characteristics of the Characteristics of donor nucleic In vitro In vivo vector acid (transgene) Regulations Regulations Regulations Regulations as amended as amended as amended as amended July 2007 Sept 2011* July 2007 Sept 2011* Any virus which meets the criteria of a Risk Group 4 Not Not microorganism in AS/NZS 2243.3:2010 with any genetic differentiated DNIR 3.1(p) differentiated DNIR 3.1(p) modification from below from below Toxin or uncharacterised gene from DNIR 3.1 (a), (b) or (c) toxin producing organism Genes whose expressed products are likely to increase the capacity of DNIR DNIR DNIR DNIR Any replication competent the virus/viral vector to induce an 3.1 (g) 3.1 (h) 3.1 (g) 3.1 (h) vector autoimmune response Creates novel replication competent virus with altered host range or DNIR DNIR DNIR DNIR mode of transmission, or increased 3.1 (h) 3.1 (i) 3.1 (h) 3.1 (i) virulence, pathogenicity or transmissibility exempt exempt Not a toxin and not a pathogenic (PC2 NLRD (PC2 NLRD determinant and not an oncogenic Non-pathogenic plant 2.1 (f) 2.1 (f) modification virus if > 25L) if > 25L) PC2 NLRD Or 2.1 (c) Exempt (PC2 Baculovirus (Autographa PC1 NLRD Oncogenic modification NLRD 2.1 (f) californica nuclear 1.1(b) polyhedrosis virus), if > 25L) polyhedrin minus PC2 NLRD -

Viral Vectors
OCCUPATIONAL HEALTH CONSIDERATIONS FOR WORK WITH LENTIVIRAL VECTORS GARY R. FUJIMOTO, M.D. OCCUPATIONAL MEDICINE CONSULTANT OCTOBER 6, 2014 Disclosure: Lecture includes off-label use of antiretroviral medications VIRAL VECTORS Definition: Viruses engineered to deliver foreign genetic material (transgene) to cells Many viral vectors deliver the genetic material into the host cells but not into the host genome where the virus replicates (unless replication incompetent) Retroviral and lentiviral vectors deliver genetic transgenes into the host chromosomes LENTIVIRAL VECTORS Human immunodeficiency virus (HIV) is a lentivirus that infects both dividing and non-dividing cells Use of the HIV virus as a viral vector has required the reengineering of the virus to achieve safe gene transfer Since HIV normally targets CD4 cells, replacing the HIV envelope gene with vesicular stomatitis virus glycoprotein (VSV-G) expands the infectious range of the vector and modes of transmission LENTIVIRAL VECTORS Remember: replication deficient lentiviral vectors integrate the vector into the host chromosomes 3rd and 4th generation constructs unlikely to become replication competent with enhanced safety due to self- inactivating vectors (however, consider present or future HIV infection) Replication deficient lentiviral vectors should be regarded as single-event infectious agents Many researchers regard these agents as relatively benign although transgene integration does occur with generally unknown effects LENTIVIRAL OCCUPATIONAL EXPOSURES Lentiviral -

Viral Vectors
505 Oldham Court Lexington, KY 40502 Phone: (859) 257-1049 Fax: (859) 323-3838 E-Mail: [email protected] http://ehs.uky.edu/biosafety VIRAL VECTORS WHAT IS A VIRAL VECTOR? Viral vectors work like a “nanosyringe” to deliver nucleic acid to a target. They are often more efficient than other transfection methods, are useful for whole organism studies, have a relatively low toxicity, and are a likely route for human gene transfer. All viral vectors require a host for replication. The production of a viral vector is typically separated from the ability of the viral vector to infect cells. While viral vectors are not typically considered infectious agents, they do maintain their ability to “infect” cells. Viral vectors just don’t replicate (although there are some replicating viral vectors in use) under experimental conditions. An HIV- based lentiviral vector no longer possesses the ability to infect an individual with HIV, but it does maintain the ability to enter a cell and express genetic information. This is why viral vectors are useful, but also require caution. If a viral vector can transfect a human cell line on a plate, it can also transfect YOUR cells if accidentally exposed. SAFETY CONSIDERATIONS FOR ALL VIRAL VECTORS When utilizing ANY viral vector, the following questions must be addressed… 1. What potential does your method of viral vector production have to generate a replication competent virus? a. Generation of viral vector refers to the number of recombination events required to form a replication competent virus. For example, if you’re using a lentivirus that is split up between 4 plasmids (gag/pol, VSV-g, rev, transgene), 3 recombination events must take place to create a replication competent virus, therefore you are using a 3rd generation lentiviral vector. -

RNA Viruses As Tools in Gene Therapy and Vaccine Development
G C A T T A C G G C A T genes Review RNA Viruses as Tools in Gene Therapy and Vaccine Development Kenneth Lundstrom PanTherapeutics, Rte de Lavaux 49, CH1095 Lutry, Switzerland; [email protected]; Tel.: +41-79-776-6351 Received: 31 January 2019; Accepted: 21 February 2019; Published: 1 March 2019 Abstract: RNA viruses have been subjected to substantial engineering efforts to support gene therapy applications and vaccine development. Typically, retroviruses, lentiviruses, alphaviruses, flaviviruses rhabdoviruses, measles viruses, Newcastle disease viruses, and picornaviruses have been employed as expression vectors for treatment of various diseases including different types of cancers, hemophilia, and infectious diseases. Moreover, vaccination with viral vectors has evaluated immunogenicity against infectious agents and protection against challenges with pathogenic organisms. Several preclinical studies in animal models have confirmed both immune responses and protection against lethal challenges. Similarly, administration of RNA viral vectors in animals implanted with tumor xenografts resulted in tumor regression and prolonged survival, and in some cases complete tumor clearance. Based on preclinical results, clinical trials have been conducted to establish the safety of RNA virus delivery. Moreover, stem cell-based lentiviral therapy provided life-long production of factor VIII potentially generating a cure for hemophilia A. Several clinical trials on cancer patients have generated anti-tumor activity, prolonged survival, and -

Lentivirus and Lentiviral Vectors Fact Sheet
Lentivirus and Lentiviral Vectors Family: Retroviridae Genus: Lentivirus Enveloped Size: ~ 80 - 120 nm in diameter Genome: Two copies of positive-sense ssRNA inside a conical capsid Risk Group: 2 Lentivirus Characteristics Lentivirus (lente-, latin for “slow”) is a group of retroviruses characterized for a long incubation period. They are classified into five serogroups according to the vertebrate hosts they infect: bovine, equine, feline, ovine/caprine and primate. Some examples of lentiviruses are Human (HIV), Simian (SIV) and Feline (FIV) Immunodeficiency Viruses. Lentiviruses can deliver large amounts of genetic information into the DNA of host cells and can integrate in both dividing and non- dividing cells. The viral genome is passed onto daughter cells during division, making it one of the most efficient gene delivery vectors. Most lentiviral vectors are based on the Human Immunodeficiency Virus (HIV), which will be used as a model of lentiviral vector in this fact sheet. Structure of the HIV Virus The structure of HIV is different from that of other retroviruses. HIV is roughly spherical with a diameter of ~120 nm. HIV is composed of two copies of positive ssRNA that code for nine genes enclosed by a conical capsid containing 2,000 copies of the p24 protein. The ssRNA is tightly bound to nucleocapsid proteins, p7, and enzymes needed for the development of the virion: reverse transcriptase (RT), proteases (PR), ribonuclease and integrase (IN). A matrix composed of p17 surrounds the capsid ensuring the integrity of the virion. This, in turn, is surrounded by an envelope composed of two layers of phospholipids taken from the membrane of a human cell when a newly formed virus particle buds from the cell. -

Viral Vectors Applied for Rnai-Based Antiviral Therapy
viruses Review Viral Vectors Applied for RNAi-Based Antiviral Therapy Kenneth Lundstrom PanTherapeutics, CH1095 Lutry, Switzerland; [email protected] Received: 30 July 2020; Accepted: 21 August 2020; Published: 23 August 2020 Abstract: RNA interference (RNAi) provides the means for alternative antiviral therapy. Delivery of RNAi in the form of short interfering RNA (siRNA), short hairpin RNA (shRNA) and micro-RNA (miRNA) have demonstrated efficacy in gene silencing for therapeutic applications against viral diseases. Bioinformatics has played an important role in the design of efficient RNAi sequences targeting various pathogenic viruses. However, stability and delivery of RNAi molecules have presented serious obstacles for reaching therapeutic efficacy. For this reason, RNA modifications and formulation of nanoparticles have proven useful for non-viral delivery of RNAi molecules. On the other hand, utilization of viral vectors and particularly self-replicating RNA virus vectors can be considered as an attractive alternative. In this review, examples of antiviral therapy applying RNAi-based approaches in various animal models will be described. Due to the current coronavirus pandemic, a special emphasis will be dedicated to targeting Coronavirus Disease-19 (COVID-19). Keywords: RNA interference; shRNA; siRNA; miRNA; gene silencing; viral vectors; RNA replicons; COVID-19 1. Introduction Since idoxuridine, the first anti-herpesvirus antiviral drug, reached the market in 1963 more than one hundred antiviral drugs have been formally approved [1]. Despite that, there is a serious need for development of novel, more efficient antiviral therapies, including drugs and vaccines, which has become even more evident all around the world today due to the recent coronavirus pandemic [2]. -

Manufacturing of Gene Therapies: Ensuring Product Safety and Quality
Manufacturing of Gene Therapies: Ensuring Product Safety and Quality SLIDE 1 This presentation will discuss the manufacturing of gene therapy products to ensure product safety and quality. SLIDE 2 This slide shows the FDA's published definition of gene therapy products. Gene therapy products are defined as all products that mediate their effects by transcription and/or translation of transferred genetic material, and/or by integrating into the host genome, and that are administered as nucleic acids, plasmids, viruses, such as adenoviral vectors, or genetically engineered micro- organisms, such as bacteria. These products can be used to modify cells in vivo, by a direct administration of a gene therapy product to a patient, or by transfer to cells ex vivo prior to patient administration. The definition shown here comes from the FDA 2006 guidance on long-term follow-up. SLIDE 3 This chart gives you an idea of the number and type of active gene therapy Investigational New Drug, or IND, applications as of July 2009. Depicted here are primarily replication defective gene therapy products, the most common being plasmid-based vectors. Plasmids can be delivered as naked DNA or they may be encapsulated in a liposome for enhanced delivery. In terms of viral vectors, adenoviral products are the most commonly seen vector. They are primarily used in cancer indications. Retroviruses have the inherent property of integrating into the host's genome and allow for stable and long-term integration of therapeutic genes. AAV stands for adeno-associated virus. AAV-vectors do not integrate into host genomes but they do allow for long-term gene expression in non-dividing cells. -

ICH Considerations on Viral/Vector Shedding; and Overview of Gene Therapy Activity in Canada
ICH Considerations on Viral/Vector Shedding; and Overview of Gene Therapy Activity in Canada Anthony Ridgway, Ph.D. Senior Regulatory Scientist Biologics & Genetic Therapies Directorate Health Canada Open Workshop on Gene Therapy Yokohama, June 12, 2009 Regulatory Authority for Gene Therapy z meets F&D Act definition of drug z satisfies Schedule D to the Act as: "Drugs obtained by rDNA procedures" "Drugs, other than antibiotics, prepared from micro-organisms" "Immunizing agents" (there are no specific regulations) (there are no gaps in regulation) Regulatory Responsibility Government of Canada Health Canada Health Products and Food Branch Biologics and Genetic Therapies Directorate Biologics and Radiopharmaceuticals Evaluation Centre Biotherapeutics Division & Clinical Evaluation Division Scientific Issues I z Route of administration; intralesional, systemic, intra-vascular (organ-targeted) z Replication competence of virus vectors; pathogenicity z Vector integration into host genome z Vector dose z Expressed product dose Scientific Issues II z Frequent lack of appropriate animal models z Choice of toxicologic investigations; (revealing unanticipated toxicities ?) z Targeting specificity z Regulation of gene expression z Immunogenicity z Contamination of drug product Safety & Ethics Considerations z Informed Consent ¾ full disclosure of risk z Institutional Review is critical ¾ protocols and consent forms z Alteration of host genome ¾ 'contamination' of non-target tissues, especially germ cells z Vector “shedding” and inadvertent generation -

Viral Vectors 101 a Desktop Resource
Viral Vectors 101 A Desktop Resource Created and Compiled by Addgene www.addgene.org August 2018 (1st Edition) Viral Vectors 101: A Desktop Resource (1st Edition) Viral Vectors 101: A desktop resource This page intentionally left blank. 2 Chapter 1 - What Are Fluorescent Proteins? ViralViral Vectors Vector 101: A Desktop Resource (1st Edition) ViralTHE VectorsHISTORY 101: OFIntroduction FLUORESCENT to this desktop PROTEINS resource (CONT’D)By Tyler J. Ford | July 16, 2018 Dear Reader, If you’ve worked with mammalian cells, it’s likely that you’ve worked with viral vectors. Viral vectors are engineered forms of mammalian viruses that make use of natural viral gene delivery machineries and that are optimized for safety and delivery. These incredibly useful tools enable you to easily deliver genes to mammalian cells and to control gene expression in a variety of ways. Addgene has been distributing viral vectors since nearly its inception in 2004. Since then, our viral Cummulative ready-to-use virus distribution through June 2018. vector collection has grown to include retroviral vectors, lentiviral vectors, adenoviral vectors, and adeno-associated viral vectors. To further enable researchers, we started our viral service in 2017. Through this service, we distribute ready-to- use, quality-controlled AAV and lentivirus for direct use in experiments. As you can see in the chart to the left, this service is already very popular and its use has grown exponentially. With this Viral Vectors 101 eBook, we are proud to further expand our viral vector offerings. Within it, you’ll find nearly all of our viral vector educational content in a single downloadable resource.