Viral Vectors 101 a Desktop Resource
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mobile Genetic Elements in Streptococci
Curr. Issues Mol. Biol. (2019) 32: 123-166. DOI: https://dx.doi.org/10.21775/cimb.032.123 Mobile Genetic Elements in Streptococci Miao Lu#, Tao Gong#, Anqi Zhang, Boyu Tang, Jiamin Chen, Zhong Zhang, Yuqing Li*, Xuedong Zhou* State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, PR China. #Miao Lu and Tao Gong contributed equally to this work. *Address correspondence to: [email protected], [email protected] Abstract Streptococci are a group of Gram-positive bacteria belonging to the family Streptococcaceae, which are responsible of multiple diseases. Some of these species can cause invasive infection that may result in life-threatening illness. Moreover, antibiotic-resistant bacteria are considerably increasing, thus imposing a global consideration. One of the main causes of this resistance is the horizontal gene transfer (HGT), associated to gene transfer agents including transposons, integrons, plasmids and bacteriophages. These agents, which are called mobile genetic elements (MGEs), encode proteins able to mediate DNA movements. This review briefly describes MGEs in streptococci, focusing on their structure and properties related to HGT and antibiotic resistance. caister.com/cimb 123 Curr. Issues Mol. Biol. (2019) Vol. 32 Mobile Genetic Elements Lu et al Introduction Streptococci are a group of Gram-positive bacteria widely distributed across human and animals. Unlike the Staphylococcus species, streptococci are catalase negative and are subclassified into the three subspecies alpha, beta and gamma according to the partial, complete or absent hemolysis induced, respectively. The beta hemolytic streptococci species are further classified by the cell wall carbohydrate composition (Lancefield, 1933) and according to human diseases in Lancefield groups A, B, C and G. -

Gutless Adenovirus: Last-Generation Adenovirus for Gene Therapy
Gene Therapy (2005) 12, S18–S27 & 2005 Nature Publishing Group All rights reserved 0969-7128/05 $30.00 www.nature.com/gt CONFERENCE PAPER Gutless adenovirus: last-generation adenovirus for gene therapy R Alba1, A Bosch1 and M Chillon1,2 1Gene Therapy Laboratory, Department of Biochemistry and Molecular Biology, Center of Animal Biotechnology and Gene Therapy (CBATEG), Universitat Auto`noma de Barcelona, Bellaterra, Spain; and 2Institut Catala` de Recerca i Estudis Avanc¸ats (ICREA), Barcelona, Spain Last-generation adenovirus vectors, also called helper-depen- viral coding regions, gutless vectors require viral proteins dent or gutless adenovirus, are very attractive for gene therapy supplied in trans by a helper virus. To remove contamination because the associated in vivo immune response is highly by a helper virus from the final preparation, different systems reduced compared to first- and second-generation adenovirus based on the excision of the helper-packaging signal have vectors, while maintaining high transduction efficiency and been generated. Among them, Cre-loxP system is mostly tropism. Nowadays, gutless adenovirus is administered in used, although contamination levels still are 0.1–1% too high different organs, such as the liver, muscle or the central to be used in clinical trials. Recently developed strategies to nervous system achieving high-level and long-term transgene avoid/reduce helper contamination were reviewed. expression in rodents and primates. However, as devoid of all Gene Therapy (2005) 12, S18–S27. doi:10.1038/sj.gt.3302612 Keywords: adenovirus; gutless; helper-dependent vectors; in vivo gene therapy Introduction clinical for more information). Nowadays, adenovirus vectors are applied to treat cancer, monogenic disorders, Gene therapy for most genetic diseases requires expres- vascular diseases and others complications. -

Viral Vectors and Biological Safety
Viral Vectors and Biological Safety Viral vectors are often designed so that they can enter human cells and deliver genes of interest. Viral vectors are usually replication-deficient – genes necessary for replication of the virus are removed from the vector and supplied separately through plasmids, helper virus, or packaging cell lines. There are several biosafety concerns that arise with the use of viral vectors including: 1) Tropism (host range) – viral vectors that can enter (infect) human cells are often used. 2) Replication-deficient viral vectors can gain back the deleted genes required for replication (become replication-competent) through recombination – referred to as replication-competent virus (RCV) breakthroughs. 3) Genes may be expressed in tissues and/or organisms where they are normally not expressed. In the case of some genes such as oncogenes, this could have far-reaching negative consequences. When evaluating safety for use of viral vectors, a number of factors need to be considered including: Risk Group (RG) of the organism; tropism (organism and tissue); route of transmission; whether the virus integrates into the host genome; and the specific gene(s) being introduced. Please contact the Office of Biological Safety (OBS) for more information on physical barriers and safety practices to use with specific viral vectors. This article concentrates on biological barriers that can be employed to improve safety when using viral vectors. Viral vectors frequently used are: • Retrovirus/lentivirus • Adenovirus • Adeno-associated virus (AAV) • Poxvirus • Herpes virus • Alphavirus • Baculovirus Amphotropic murine leukemia virus (MLV) – also called Moloney murine leukemia virus (MMLV) – and adenovirus are common viral vectors used to introduce genes into human cells. -

Neutralizing Antibody Against SARS-Cov-2 Spike in COVID-19 Patients, Health Care Workers, and Convalescent Plasma Donors
TECHNICAL ADVANCE Neutralizing antibody against SARS-CoV-2 spike in COVID-19 patients, health care workers, and convalescent plasma donors Cong Zeng,1,2 John P. Evans,1,2,3 Rebecca Pearson,4 Panke Qu,1,2 Yi-Min Zheng,1,2 Richard T. Robinson,5 Luanne Hall-Stoodley,5 Jacob Yount,5 Sonal Pannu,6 Rama K. Mallampalli,6 Linda Saif,7,8 Eugene Oltz,5 Gerard Lozanski,4 and Shan-Lu Liu1,2,5,8 1Center for Retrovirus Research, 2Department of Veterinary Biosciences, 3Molecular, Cellular and Developmental Biology Program, 4Department of Pathology, 5Department of Microbial Infection and Immunity, and 6Department of Medicine, The Ohio State University (OSU), Columbus, Ohio, USA. 7Food Animal Health Research Program, Ohio Agricultural Research and Development Center, Wooster, Ohio, USA. 8Viruses and Emerging Pathogens Program, Infectious Diseases Institute, OSU, Columbus, Ohio, USA. Rapid and specific antibody testing is crucial for improved understanding, control, and treatment of COVID-19 pathogenesis. Herein, we describe and apply a rapid, sensitive, and accurate virus neutralization assay for SARS-CoV-2 antibodies. The assay is based on an HIV-1 lentiviral vector that contains a secreted intron Gaussia luciferase (Gluc) or secreted nano-luciferase reporter cassette, pseudotyped with the SARS-CoV-2 spike (S) glycoprotein, and is validated with a plaque-reduction assay using an authentic, infectious SARS-CoV-2 strain. The assay was used to evaluate SARS-CoV-2 antibodies in serum from individuals with a broad range of COVID-19 symptoms; patients included those in the intensive care unit (ICU), health care workers (HCWs), and convalescent plasma donors. -
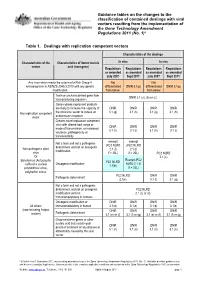
Guidance Tables on the Changes To
Guidance tables on the changes to the classification of contained dealings with viral vectors resulting from the implementation of the Gene Technology Amendment Regulations 2011 (No. 1)* Table 1. Dealings with replication competent vectors Characteristics of the dealings Characteristics of the Characteristics of donor nucleic In vitro In vivo vector acid (transgene) Regulations Regulations Regulations Regulations as amended as amended as amended as amended July 2007 Sept 2011* July 2007 Sept 2011* Any virus which meets the criteria of a Risk Group 4 Not Not microorganism in AS/NZS 2243.3:2010 with any genetic differentiated DNIR 3.1(p) differentiated DNIR 3.1(p) modification from below from below Toxin or uncharacterised gene from DNIR 3.1 (a), (b) or (c) toxin producing organism Genes whose expressed products are likely to increase the capacity of DNIR DNIR DNIR DNIR Any replication competent the virus/viral vector to induce an 3.1 (g) 3.1 (h) 3.1 (g) 3.1 (h) vector autoimmune response Creates novel replication competent virus with altered host range or DNIR DNIR DNIR DNIR mode of transmission, or increased 3.1 (h) 3.1 (i) 3.1 (h) 3.1 (i) virulence, pathogenicity or transmissibility exempt exempt Not a toxin and not a pathogenic (PC2 NLRD (PC2 NLRD determinant and not an oncogenic Non-pathogenic plant 2.1 (f) 2.1 (f) modification virus if > 25L) if > 25L) PC2 NLRD Or 2.1 (c) Exempt (PC2 Baculovirus (Autographa PC1 NLRD Oncogenic modification NLRD 2.1 (f) californica nuclear 1.1(b) polyhedrosis virus), if > 25L) polyhedrin minus PC2 NLRD -

DNA Recombination Is Sufficient for Retroviral Transduction JODY R
Proc. Natl. Acad. Sci. USA Vol. 92, pp. 2460-2464, March 1995 Biochemistry DNA recombination is sufficient for retroviral transduction JODY R. SCHWARTZ, Susi DUESBERG, AND PETER H. DUESBERG Department of Molecular and Cell Biology, University of California, Berkeley, Berkeley, CA 94720-3206 Contributed by Peter H. Duesberg, November 28, 1994 ABSTRACT Oncogenic retroviruses carry coding se- quences that are transduced from cellular protooncogenes. Nat- env pOl X ural transduction involves two nonhomologous recombinations and is thus extremely rare. Since transduction has never been reproduced experimentall, its mechanism has been studied in terms oftwoehypotheses: (i) the DNAmodel,which postulates two ___ _-I ____ DNA recombinations, and (ii) the RNA model, which postulates a 5' DNA recombination and a 3' RNA recombination occurring during reverse transcription of viral and protooncogene RNA. proto-onc gene Here we use two viral DNA constructs to test the prediction ofthe DNA model that the 3' DNA recombination is achieved by conventional integration of a retroviral DNA 3' of the chromo- somal protooncogene coding region. For the DNA model to be ga-onc - gag viable, such recombinant viruses must be infectious without the w5 essential tract that precedes the 3' purportedly polypurine (ppt) FIG. 1. The DNA model of retroviral transduction. The model long terminal repeat (LTR) of all retroviruses. Our constructs proposes that the 5' retrovirus/protooncogene junction is achieved by consist ofa ras coding region from Harvey sarcoma virus which nonhomologous recombination between a circular provirus with a is naturally linked at the 5' end to a retroviral LTR and single LTR. Such proviruses are common in virus-infected cells (1). -

SARS-Cov-2 RBD Antibodies That Maximize Breadth and Resistance to Escape W 1,15 2,15 3,15 4 Tyler N
https://doi.org/10.1038/s41586-021-03807-6 Accelerated Article Preview SARS-CoV-2 RBD antibodies that maximize W breadth and resistance to escape E VI Received: 29 March 2021 Tyler N. Starr, N ad in e Czudnochowski, Zhuoming Liu, Fabrizia Zatta, Young-Jun Park, Amin Addetia, Dora Pinto, Martina Beltramello, Patrick Hernandez, AllisonE J. Greaney, Accepted: 6 July 2021 Roberta Marzi, William G. Glass, Ivy Zhang, Adam S. Dingens, John E. B ow en, Accelerated Article Preview Published M . Alejandra Tortorici, Alexandra C. W al ls , J as on A. Wojcechowskyj,R Anna De Marco, online 14 July 2021 Laura E. Rosen, Jiayi Zhou, Martin Montiel-Ruiz, Hannah Kaiser, Josh Dillen, Heather Tucker, Jessica Bassi, Chiara Silacci-Fregni, Michael P. Housley,P Julia di Iulio, Gloria Lombardo, Cite this article as: Starr, T. N. et al. Maria Agostini, Nicole Sprugasci, Katja Culap, Stefano Jaconi, Marcel Meury, SARS-CoV-2 RBD antibodies that maximize Exequiel Dellota, Rana Abdelnabi, Shi-Yan Caroline Foo, Elisabetta Cameroni, breadth and resistance to escape. Nature Spencer Stumpf, Tristan I. Croll, Jay C. Nix, ColinE Havenar-Daughton, Luca Piccoli, https://doi.org/10.1038/s41586-021-03807-6 Fabio Benigni, Johan Neyts, Amalio Telenti, Florian A. Lempp, Matteo S. Pizzuto, (2021). John D. Chodera, Christy M. Hebner, HerbertL W. Virgin, Sean P. J. Whelan, David Veesler, Davide Corti, Jesse D. Bloom & Gyorgy Snell I C This is a PDF fle of a peer-reviewed paper that has been accepted for publication. Although unedited, the Tcontent has been subjected to preliminary formatting. Nature is providing this earlyR version of the typeset paper as a service to our authors and readers. -

The LUCA and Its Complex Virome in Another Recent Synthesis, We Examined the Origins of the Replication and Structural Mart Krupovic , Valerian V
PERSPECTIVES archaea that form several distinct, seemingly unrelated groups16–18. The LUCA and its complex virome In another recent synthesis, we examined the origins of the replication and structural Mart Krupovic , Valerian V. Dolja and Eugene V. Koonin modules of viruses and posited a ‘chimeric’ scenario of virus evolution19. Under this Abstract | The last universal cellular ancestor (LUCA) is the most recent population model, the replication machineries of each of of organisms from which all cellular life on Earth descends. The reconstruction of the four realms derive from the primordial the genome and phenotype of the LUCA is a major challenge in evolutionary pool of genetic elements, whereas the major biology. Given that all life forms are associated with viruses and/or other mobile virion structural proteins were acquired genetic elements, there is no doubt that the LUCA was a host to viruses. Here, by from cellular hosts at different stages of evolution giving rise to bona fide viruses. projecting back in time using the extant distribution of viruses across the two In this Perspective article, we combine primary domains of life, bacteria and archaea, and tracing the evolutionary this recent work with observations on the histories of some key virus genes, we attempt a reconstruction of the LUCA virome. host ranges of viruses in each of the four Even a conservative version of this reconstruction suggests a remarkably complex realms, along with deeper reconstructions virome that already included the main groups of extant viruses of bacteria and of virus evolution, to tentatively infer archaea. We further present evidence of extensive virus evolution antedating the the composition of the virome of the last universal cellular ancestor (LUCA; also LUCA. -

Molecular Characterization of Human Papillomavirus Type 159 (HPV159)
viruses Article Molecular Characterization of Human Papillomavirus Type 159 (HPV159) Iva Markovi´c 1, Lea Hošnjak 2 , Katja Seme 2 and Mario Poljak 2,* 1 Division of Molecular Biology, Ruder¯ Boškovi´cInstitute, BijeniˇckaCesta 54, 10000 Zagreb, Croatia; [email protected] 2 Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška Cesta 4, 1105 Ljubljana, Slovenia; [email protected] (L.H.); [email protected] (K.S.) * Correspondence: [email protected]; Tel.: +386-1-543-7453 Abstract: Human papillomavirus type 159 (HPV159) was identified in an anal swab sample and preliminarily genetically characterized by our group in 2012. Here we present a detailed molecular in silico analysis that showed that the HPV159 viral genome is 7443 bp in length and divided into five early and two late genes, with conserved functional domains and motifs, and a non-coding long control region (LCR) with significant regulatory sequences that allow the virus to complete its life cycle and infect novel host cells. HPV159, clustering into the cutaneotropic Betapapillomavirus (Beta-PV) genus, is phylogenetically most similar to HPV9, forming an individual phylogenetic group in the viral species Beta-2. After testing a large representative collection of clinical samples with HPV159 type-specific RT-PCR, in addition to the anal canal from which the first HPV159 isolate was obtained, HPV159 was further detected in other muco-cutaneous (4/181, 2.2%), mucosal (22/764, 2.9%), and cutaneous (14/554, 2.5%) clinical samples, suggesting its extensive tissue tropism. However, because very low HPV159 viral loads were estimated in the majority of positive samples, Citation: Markovi´c,I.; Hošnjak, L.; it seemed that HPV159 mainly caused clinically insignificant infections of the skin and mucosa. -

Virus Goes Viral: an Educational Kit for Virology Classes
Souza et al. Virology Journal (2020) 17:13 https://doi.org/10.1186/s12985-020-1291-9 RESEARCH Open Access Virus goes viral: an educational kit for virology classes Gabriel Augusto Pires de Souza1†, Victória Fulgêncio Queiroz1†, Maurício Teixeira Lima1†, Erik Vinicius de Sousa Reis1, Luiz Felipe Leomil Coelho2 and Jônatas Santos Abrahão1* Abstract Background: Viruses are the most numerous entities on Earth and have also been central to many episodes in the history of humankind. As the study of viruses progresses further and further, there are several limitations in transferring this knowledge to undergraduate and high school students. This deficiency is due to the difficulty in designing hands-on lessons that allow students to better absorb content, given limited financial resources and facilities, as well as the difficulty of exploiting viral particles, due to their small dimensions. The development of tools for teaching virology is important to encourage educators to expand on the covered topics and connect them to recent findings. Discoveries, such as giant DNA viruses, have provided an opportunity to explore aspects of viral particles in ways never seen before. Coupling these novel findings with techniques already explored by classical virology, including visualization of cytopathic effects on permissive cells, may represent a new way for teaching virology. This work aimed to develop a slide microscope kit that explores giant virus particles and some aspects of animal virus interaction with cell lines, with the goal of providing an innovative approach to virology teaching. Methods: Slides were produced by staining, with crystal violet, purified giant viruses and BSC-40 and Vero cells infected with viruses of the genera Orthopoxvirus, Flavivirus, and Alphavirus. -

Spread of a SARS-Cov-2 Variant Through Europe in the Summer of 2020
Article Spread of a SARS-CoV-2 variant through Europe in the summer of 2020 https://doi.org/10.1038/s41586-021-03677-y Emma B. Hodcroft1,2,3 ✉, Moira Zuber1, Sarah Nadeau2,4, Timothy G. Vaughan2,4, Katharine H. D. Crawford5,6,7, Christian L. Althaus3, Martina L. Reichmuth3, John E. Bowen8, Received: 25 November 2020 Alexandra C. Walls8, Davide Corti9, Jesse D. Bloom5,6,10, David Veesler8, David Mateo11, Accepted: 28 May 2021 Alberto Hernando11, Iñaki Comas12,13, Fernando González-Candelas13,14, SeqCOVID-SPAIN consortium*, Tanja Stadler2,4,92 & Richard A. Neher1,2,92 ✉ Published online: 7 June 2021 Check for updates Following its emergence in late 2019, the spread of SARS-CoV-21,2 has been tracked by phylogenetic analysis of viral genome sequences in unprecedented detail3–5. Although the virus spread globally in early 2020 before borders closed, intercontinental travel has since been greatly reduced. However, travel within Europe resumed in the summer of 2020. Here we report on a SARS-CoV-2 variant, 20E (EU1), that was identifed in Spain in early summer 2020 and subsequently spread across Europe. We fnd no evidence that this variant has increased transmissibility, but instead demonstrate how rising incidence in Spain, resumption of travel, and lack of efective screening and containment may explain the variant’s success. Despite travel restrictions, we estimate that 20E (EU1) was introduced hundreds of times to European countries by summertime travellers, which is likely to have undermined local eforts to minimize infection with SARS-CoV-2. Our results illustrate how a variant can rapidly become dominant even in the absence of a substantial transmission advantage in favourable epidemiological settings. -

Effects of Retroviruses on Host Genome Function
ANRV361-GE42-20 ARI 1 August 2008 18:2 V I E E W R S I E N C N A D V A Effects of Retroviruses on Host Genome Function Patric Jern and John M. Coffin Department of Molecular Biology and Microbiology, Tufts University School of Medicine, Boston, Massachusetts 02111; email: [email protected], John.Coffi[email protected] Annu. Rev. Genet. 2008. 42:20.1–20.23 Key Words The Annual Review of Genetics is online at Human Endogenous Retrovirus, LTR, transcription, recombination, genet.annualreviews.org methylation This article’s doi: 10.1146/annurev.genet.42.110807.091501 Abstract Copyright c 2008 by Annual Reviews. For millions of years, retroviral infections have challenged vertebrates, All rights reserved occasionally leading to germline integration and inheritance as ERVs, 0066-4197/08/1201-0001$20.00 genetic parasites whose remnants today constitute some 7% to 8% of the human genome. Although they have had significant evolutionary side effects, it is useful to view ERVs as fossil representatives of retro- viruses extant at the time of their insertion into the germline, not as direct players in the evolutionary process itself. Expression of particu- lar ERVs is associated with several positive physiological functions as well as certain diseases, although their roles in human disease as etio- logical agents, possible contributing factors, or disease markers—well demonstrated in animal models—remain to be established. Here we discuss ERV contributions to host genome structure and function, in- cluding their ability to mediate recombination, and physiological effects on the host transcriptome resulting from their integration, expression, and other events.