The Alkylation of the Boron Hydrides, Reaction of Pentaborane-9 with 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Synthesis of Bromochloromethane Using Phase Transfer Catalysis
1 SYNTHESIS OF BROMOCHLOROMETHANE USING PHASE TRANSFER CATALYSIS By LANCELOT LUCRETIUS BROOKS Baccalaureus Scientiae Honores-Chemistry, Nelson Mandela Metropolitan University A dissertation submitted in fulfillment of the requirements for the Masters Degree in Chemistry In the Faculty of Science at the NELSON MANDELA METROPOLITAN UNIVERSITY Nov. 2011 Promoter : Dr G. Dugmore Co-Promoter : Prof B. Zeelie 2 DECLARATION I, Lancelot Brooks, hereby declare that the above-mentioned treatise is my own work and that it has not previously been submitted for assessment to another University, or for another qualification. ……………………………….. ……………………….. Mr. L.L. Brooks Date 3 ACKNOWLEDGEMENTS To my promoters Dr. Gary Dugmore, and Prof. Ben Zeelie for their invaluable input, help and guidance. To NRF and NMMU for financial assistance To my parents and brothers for their love and support To Peter, Batsho, Unati, and friends in NMMU chemistry research laboratory, thank you guys. To my dearest fiancée, Natasha, a very special thank you for always being there and supporting me. Love you angel. “And we know that all things work together for good to those who love God, to those who are called according to His purpose” -Romans 8:28. 4 TABLE OF CONTENTS DECLARATION……………………………………………………………………. 2 ACKNOWLEDGEMENTS……………………………………………………………. 3 TABLE OF CONTENTS………………………………………………………………. 4 LIST OF FIGURES…………………………………………………………………….. 8 LIST OF TABLES……………………………………………………………………… 9 LIST OF EQUATIONS………………………………………………………………… 11 SUMMARY……………………………………………………………………………… 12 CHAPTER 1…………………………………………………………………………….. 14 INTRODUCTION………………………………………………………………………. 14 1.1. Technology of leather production……………………………………………….. 14 1.2. Synthesis of TCMTB……………………………………………………………… 17 1.3. Bromine……………………………………………………………………………. 20 1.3.1. Overview……………………………………………………………. 20 1.3.2. Applications of bromine compounds…………..…………………. 22 1.3.2.1. Photography……………………………………………… 22 1.3.2.2. -

Supplement to the Role of Internal Standards and Their Interaction With
Supplement to: Hiatt, M., “The Role of Internal Standards and their Interaction with Soils Impact Accuracy of Volatile Organics Determinations,” Int. J. of Environ. Anal. Chem., 2010 90:8 591-604. Please note that this is a non-copyrighted web version of supplemental information belonging to the above-cited journal article. There are formatting and page-numbering differences between this web version and the actual published version. There are also minor content differences. This version can be accessed at: http://www.epa.gov/nerlesd1/chemistry/vacuum/reference/pubs.htm This publication is also available from Informaworld: http://www.informaworld.com/smpp/content~db=all~content=a921288588 Web version, 27 April 2010 1 Supplement to: The Role of Internal Standards and their Interaction with Soils Impact Accuracy of Volatile Organics Determinations Michael H. Hiatt U.S. Environmental Protection Agency, National Exposure Research Laboratory Environmental Sciences Division. P.O. Box 93478, Las Vegas, Nevada 89193-3478 Phone: 702 798 2381. Fax: 702 798 2142. E-mail: [email protected]. Supplementary Information Data that was too detailed for “The Role of Internal Standards and their Interaction with Soils Impact Accuracy of Volatile Organics Determinations” is presented as supplemental information. This information includes each analyte by matrix and includes the accuracy evaluation, analyte results impacted by criteria, and results from varying spike equilibration times. The list of internal standards and their chemical properties are presented in Table S1 while the surrogates are listed in Table S2. Tables of results by analyte are included as Tables S3-8. These tables include the data by matrix and combined. -

SROC Annex V
Annex V Major Chemical Formulae and Nomenclature This annex presents the formulae and nomenclature for halogen-containing species and other species that are referred to in this report (Annex V.1). The nomenclature for refrigerants and refrigerant blends is given in Annex V.2. V.1 Substances by Groupings V.1.1 Halogen-Containing Species V.1.1.1 Inorganic Halogen-Containing Species Atomic chlorine Cl Atomic bromine Br Molecular chlorine Cl2 Molecular bromine Br2 Chlorine monoxide ClO Bromine monoxide BrO Chlorine radicals ClOx Bromine radicals BrOx Chloroperoxy radical ClOO Bromine nitrate BrONO2, BrNO3 Dichlorine peroxide (ClO dimer) (ClO)2, Cl2O2 Potassium bromide KBr Hydrogen chloride (Hydrochloric acid) HCl Inorganic chlorine Cly Antimony pentachloride SbCl5 Atomic fluorine F Molecular fluorine F2 Atomic iodine I Hydrogen fluoride (Hydrofluoric acid) HF Molecular iodine I2 Sulphur hexafluoride SF6 Nitrogen trifluoride NF3 IPCC Boek (dik).indb 467 15-08-2005 10:57:13 468 IPCC/TEAP Special Report: Safeguarding the Ozone Layer and the Global Climate System V.1.1.2 Halocarbons For each halocarbon the following information is given in columns: • Chemical compound [Number of isomers]1 (or common name) • Chemical formula • CAS number2 • Chemical name (or alternative name) V.1.1.2.1 Chlorofluorocarbons (CFCs) CFC-11 CCl3F 75-69-4 Trichlorofluoromethane CFC-12 CCl2F2 75-71-8 Dichlorodifluoromethane CFC-13 CClF3 75-72-9 Chlorotrifluoromethane CFC-113 [2] C2Cl3F3 Trichlorotrifluoroethane CCl FCClF 76-13-1 CFC-113 2 2 1,1,2-Trichloro-1,2,2-trifluoroethane -

ACE Tips for Filing EPA ODS
Automated Commercial Environment ACE Tips for Filing EPA ODS June, 2018 Ozone-depleting substances (ODS) deplete the stratospheric ozone layer when the chlorine and bromine atoms that they contain come into contact with ozone molecules. One chlorine atom can destroy over 100,000 ozone molecules before it is removed from the stratosphere. ODS that release chlorine include chlorofluorocarbons (CFCs), hydrochlorofluorocarbons (HCFCs), carbon tetrachloride, and methyl chloroform. ODS that release bromine include halons and methyl bromide. HTS code Ozone-Depleting Substance 2903.14.0000 Carbon tetrachloride What commodities 2903.19.6010 Methylchloroform (1,1,1-Trichloroethane) are ODS used in? 2903.39.1520 Methyl bromide 2903.71.0000 Chlorodifluoromethane (HCFC-22) ODS have been used 2903.72.0020 Dichlorotrifluoroethane (HCFC-123) for refrigeration, air 2903.73.0000 Dichlorofluoroethanes (HCFC-141, 141b) conditioning, insulation, 2903.74.0000 Chlorodifluoroethanes (HCFC-142, 142b) solvents, aerosol 2903.75.0000 Dichloropentafluoropropanes propellants, and in other (HCFC-225, 225ca, 225cb) sectors. Currently, the United 2903.76.0010 Bromotrifluoromethane (Halon 1301) States is in the 2903.76.0050 Bromochlorodifluoromethane (Halon 1211), process of phasing bromotrifluoromethane (Halon 1301), and out HCFCs, but in the dibromotetrafluoroethanes (Halon 2402), Other interim limited imports are 2903.77.0010 Trichlorofluoromethane (CFC-11) allowed. CFCs, 2903.77.0020 Trichlorotrifluoroethanes (CFC-113, CFC-113a) methyl bromide, halon, 2903.77.0030 -

Maine Remedial Action Guidelines (Rags) for Contaminated Sites
Maine Department of Environmental Protection Remedial Action Guidelines for Contaminated Sites (RAGs) Effective Date: May 1, 2021 Approved by: ___________________________ Date: April 27, 2021 David Burns, Director Bureau of Remediation & Waste Management Executive Summary MAINE DEPARTMENT OF ENVIRONMENTAL PROTECTION 17 State House Station | Augusta, Maine 04333-0017 www.maine.gov/dep Maine Department of Environmental Protection Remedial Action Guidelines for Contaminated Sites Contents 1 Disclaimer ...................................................................................................................... 1 2 Introduction and Purpose ............................................................................................... 1 2.1 Purpose ......................................................................................................................................... 1 2.2 Consistency with Superfund Risk Assessment .............................................................................. 1 2.3 When to Use RAGs and When to Develop a Site-Specific Risk Assessment ................................. 1 3 Applicability ................................................................................................................... 2 3.1 Applicable Programs & DEP Approval Process ............................................................................. 2 3.1.1 Uncontrolled Hazardous Substance Sites ............................................................................. 2 3.1.2 Voluntary Response Action Program -

Hal-List.Pdf
Florida Department of Health Environmental Health Bureau of Environmental Health, Water Programs Maximum Contaminant Levels and Health Advisory Levels Last Update 8/4/2016 HAL = Health Advisory Level MCL = Maximum Contaminant Level SMCL = Secondary Maximum Contaminant Level ug/L = micrograms per Liter or parts per billion mg/L = milligrams per Liter or parts per million MFL = million fibers per liter Chemical Name Units MCL/HAL 1,1,1,2‐TETRACHLOROETHANE 1 ug/L HAL ALPHA‐BHC 0.04 ug/L HAL CHLORPROPHAM 1400 ug/L HAL GLYPHOSATE 700 ug/L MCL PENTACHLOROPHENOL 1 ug/L MCL 1,1,1‐TRICHLOROETHANE 200 ug/L MCL 1,1,2,2‐TETRACHLOROETHANE 1 ug/L HAL 1,1,2‐TRICHLOROETHANE 5 ug/L MCL 1,1‐DICHLOROETHANE 70 ug/L HAL 1,1‐DICHLOROETHYLENE 7 ug/L MCL 1,2,3‐TRICHLOROBENZENE 70 ug/L HAL 1,2,3‐TRICHLOROPROPANE 40 ug/L HAL 1,2,4‐TRICHLOROBENZENE 70 ug/L MCL 1,2,4‐TRIMETHYLBENZENE 35 ug/L HAL 1,2‐DICHLOROETHANE 3 ug/L MCL 1,2‐DICHLOROPROPANE 5 ug/L MCL 1,3,5‐TRIMETHYLBENZENE 35 ug/L HAL 1,4‐DIOXANE 0.35 ug/L HAL 1‐CHLOROBUTANE 2800 ug/L HAL 2,3,7,8 TCDD (DIOXIN) 0.00003 ug/L MCL 2,4,5‐T 70 ug/L HAL 2,4,5‐TP (SILVEX) 50 ug/L MCL 2,4‐D 70 ug/L MCL 2,4‐DB 56 ug/L HAL 2,4‐DINITROTOLUENE 0.05 ug/L HAL 2,6‐DINITROTOLUENE 0.05 ug/L HAL 2‐BUTANONE 4200 ug/L HAL 2‐HEXANONE 280 ug/L HAL 4,4'‐DDD 0.1 ug/L HAL 4,4'‐DDE 0.1 ug/L HAL 4,4'‐DDT 0.1 ug/L HAL 4‐NITROPHENOL 56 ug/L HAL 8/4/2016 Chemical Name Units MCL/HAL ACENAPHTHYLENE 210 ug/L HAL ACIFLUORFEN 1 ug/L HAL ALACHLOR 2 ug/L MCL ALDICARB (SUM OF ALL ALDICARBS) 7 ug/L HAL ALDICARB SULFONE (SUM OF ALDICARBS) 7 ug/L HAL -
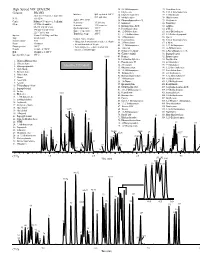
GC and GC/MS Applications Environmental-Volatiles
GC and GC/MS Applications Environmental-Volatiles High Speed VOC EPA 8260 32. 2,2-Dichloropropane 75. Tetrachloroethene Column: DB-VRX 33. Ethyl acetate 76. 1,1,1,2-Tetrachloroethane Interface: Split injector at 150°C, 34. Ethyl-tert-butyl ether 77. 1-Chlorohexane 20 m x 0.18 mm id, 1.0 µm film 60:1 split ratio P/N: 121-1524 35. Methyl acrylate 78. Chlorobenzene Agilent 5973 MSD 36. Dibromofluoromethane IS 79. Ethylbenzene Carrier: Helium at 55 cm/sec (1.5 mL/min) Scan range: 35-260 amu Oven: 45°C for 3.0 minutes 37. Isobutanol 80. Bromoform Scan rate: 3.25 scans/sec 38. Dichloroethane-d4 IS 81. m-Xylene 45-190°C at 36°C/min Quad temperature: 150°C 190-225°C at 20°C/min 39. Pentafluorobenzene 82. p-Xylene Source temperature: 200°C 40. 1,2-Dichloroethane 83. trans-Dichlorobutene 225°C for 0.5 min Transfer line temp: 200°C Injector: Tekmar 3100 Purge and Trap 41. 1,1,1-Trichloroethane 84. 1,3-Dichloro-2-propanol 42. 1-Chlorobutane 85. Styrene Trap: Vocarb 3000 Sample Concentration Sample volume: 5 mL 43. Crotonaldehyde 86. 1,1,2,2-Tetrachloroethane • Halogentated and aromatic analytes at 40 ppb 44. 2-Chloroethanol 87. o-Xylene Purge: 11 minutes • Internal standards at 20 ppb Desorb preheat: 245°C 45. 1,1-Dichloropropene 88. 1,2,3-Trichlropropane • Polar analytes (i.e., ethers, alcohols and 46. 1-Butanol 89. cis-Dichlorobutene Desorb: 1 minute at 250°C ketones at 100-800 ppb) Bake: 10 minutes at 260°C 47. -
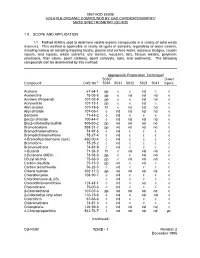
Method 8260B: Volatile Organic Compounds by Gas
METHOD 8260B VOLATILE ORGANIC COMPOUNDS BY GAS CHROMATOGRAPHY/ MASS SPECTROMETRY (GC/MS) 1.0 SCOPE AND APPLICATION 1.1 Method 8260 is used to determine volatile organic compounds in a variety of solid waste matrices. This method is applicable to nearly all types of samples, regardless of water content, including various air sampling trapping media, ground and surface water, aqueous sludges, caustic liquors, acid liquors, waste solvents, oily wastes, mousses, tars, fibrous wastes, polymeric emulsions, filter cakes, spent carbons, spent catalysts, soils, and sediments. The following compounds can be determined by this method: Appropriate Preparation Techniquea 5030/ Direct Compound CAS No.b 5035 5031 5032 5021 5041 Inject. Acetone 67-64-1 pp c c nd c c Acetonitrile 75-05-8 pp c nd nd nd c Acrolein (Propenal) 107-02-8 pp c c nd nd c Acrylonitrile 107-13-1 pp c c nd c c Allyl alcohol 107-18-6 ht c nd nd nd c Allyl chloride 107-05-1 c nd nd nd nd c Benzene 71-43-2 c nd c c c c Benzyl chloride 100-44-7 c nd nd nd nd c Bis(2-chloroethyl)sulfide 505-60-2 pp nd nd nd nd c Bromoacetone 598-31-2 pp nd nd nd nd c Bromochloromethane 74-97-5 c nd c c c c Bromodichloromethane 75-27-4 c nd c c c c 4-Bromofluorobenzene (surr) 460-00-4 c nd c c c c Bromoform 75-25-2 c nd c c c c Bromomethane 74-83-9 c nd c c c c n-Butanol 71-36-3 ht c nd nd nd c 2-Butanone (MEK) 78-93-3 pp c c nd nd c t-Butyl alcohol 75-65-0 pp c nd nd nd c Carbon disulfide 75-15-0 pp nd c nd c c Carbon tetrachloride 56-23-5 c nd c c c c Chloral hydrate 302-17-0 pp nd nd nd nd c Chlorobenzene -

Environmental Fee Schedule
BROOKSIDE LABORATORIES, INC. Environmental Fee Schedule REVISED: June 15, 2020 BROOKSIDE LABORATORIES, INC. ENVIRONMENTAL FEE SCHEDULE Code: DRINKING WATER Individual Test IA590 pH (SM 4500-H+) IA800 TOTAL DISSOLVED SOLIDS (TDS) (EPA 160.1) IA030 ALKALINITY (SM 2320-B) IA050 ALKALINITY – BICARBONATE (SM 2320-B) IA110 ALKALINITY – CARBONATE (SM 2320-B) IA033 ALKALINITY – HYDROXIDE (SM 2320-B) IA112 ALKALINITY – PHENOLPHTHALEIN (SM 2320-B) ANIONS by CHROMATOGRAPHY include: Bromide; Chloride; Fluoride; Nitrogen-Nitrate; Nitrogen-Nitrite; Sulfate IA430 NITRATE + NITRITE COMBO (EPA 300.0) IA080 BROMIDE (300.1) IA647 PHOSPHORUS – TOTAL (SM 4500P-E) IA240 CYANIDE (EPA 335.4) METALS (EPA 200.7) Aluminum, Antimony, Arsenic, Barium, Beryllium, Boron, Cadmium, Calcium, Chromium (Total), Cobalt, Copper, Hardness – Calcium, Hardness – Total, Iron, Lead, Lithium, Magnesium, Manganese, Mercury, Molybdenum, Nickel, Phosphorus, Potassium, Selenium, Silica, Silver, Sodium, Sulfur, Strontium, Thallium, Tin, Titanium, Vanadium, Zinc SEE INDIVIDUAL METALS FOR SEPARATE TEST CODES PRIMARY DRINKING WATER CONTAMINANT METALS (EPA 200.5) includes: Aluminum, Antimony, Arsenic, Barium, Beryllium, Cadmium, Chromium (Total), Copper, Iron, Lead, Manganese, Mercury, Molybdenum, Nickel, Selenium, Silver, Thallium, Vanadium, Zinc SEE INDIVIDUAL METALS FOR SEPARATE TEST CODES LEAD AND COPPER RULE includes: EE184 Lead LDL, Copper LDL IB170 MERCURY (EPA 245.1) IB261 THALLIUM (EPA 200.8) SOC’S GROUP A (EPA 525.2) includes: EW037 Alachlor; Atrazine; Hexachlorobenzene; Hexachlorocyclopentadiene; -

The Replacement of Fire Extinguishers for Military Applications
INITIAL INVESTIGATION OF COMBINED FIRE EXTINGUISHING EFFICIENCY OF NOVEL PHOSPHORUS CONTAINING COMPOUNDS IN POTENTIAL DELIVERY MEDIA. Dr Emmajane L Morrey and Linda C Knutsen QinetiQ, Fire Research & Characterisation, Haslar Marine Technology Park, Haslar Road, Gosport, Hampshire. PO12 2AG. UK. Tel: +44(0)23 9233 5353; Fax: +44(0)23 9233 5592; e-mail: [email protected] INTRODUCTION A suite of novel phosphorus containing compounds have been designed and synthesised through QinetiQ research. These have been evaluated for extinguishing concentration on the FID, a screening method of measuring extinguishing concentration to simulate the cup burner, developed by QinetiQ, and found to be very efficient extinguishing compounds [1,2,3]. Results showed that 1-2% by volume of agent was sufficient to extinguish the FID hydrocarbon / air diffusion flame, whereas 3% by volume of Halon 1301 was required. The extinguishing concentrations for some compounds previously measured are summarised in Table 1. The phosphorus compounds are liquids with boiling points between about 180-280°C, compared with Halon 1301, which has a boiling point of –57.8°C, i.e. is a gas at room temperature. This means that in order to progress the phosphorus compounds from novel chemicals towards marketable agents, a method for delivery & dispersion of the compounds needs to be developed. Table 1. Summary of extinguishing concentrations for some compounds previously reported Compound Cup Burner FID Bromotrifluoromethane (Halon 1301) 2.9 3.0 Bromochlorodifluoromethane (Halon -

Defining Potential Chemical Peaks and Management Options
PROJECT NO. 4991 Defining Potential Chemical Peaks and Management Options Defining Potential Chemical Peaks and Management Options Prepared by: Jean Debroux Kennedy Jenks Consultants Megan H. Plumlee Orange County Water District Shane Trussell Trussell Technologies, Inc. Co-sponsored by: California State Water Resources Control Board 2021 The Water Research Foundation (WRF) is a nonprofit (501c3) organization which provides a unified source for One Water research and a strong presence in relationships with partner organizations, government and regulatory agencies, and Congress. The foundation conducts research in all areas of drinking water, wastewater, stormwater, and water reuse. The Water Research Foundation’s research portfolio is valued at over $700 million. WRF plays an important role in the translation and dissemination of applied research, technology demonstration, and education, through creation of research-based educational tools and technology exchange opportunities. WRF serves as a leader and model for collaboration across the water industry and its materials are used to inform policymakers and the public on the science, economic value, and environmental benefits of using and recovering resources found in water, as well as the feasibility of implementing new technologies. For more information, contact: The Water Research Foundation 1199 North Fairfax Street, Suite 900 6666 West Quincy Avenue Alexandria, VA 22314-1445 Denver, Colorado 80235-3098 www.waterrf.org P 571.384.2100 P 303.347.6100 [email protected] ©Copyright 2021 by The Water Research Foundation. All rights reserved. Permission to copy must be obtained from The Water Research Foundation. WRF ISBN: 978-1-60573-555-9 WRF Project Number: 4991 This report was prepared by the organization(s) named below as an account of work sponsored by The Water Research Foundation.