Method 8260C: Volatile Organic Compounds by Gas
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Iodomethane Safety Data Sheet 1100H01 According to Federal Register / Vol
Iodomethane Safety Data Sheet 1100H01 according to Federal Register / Vol. 77, No. 58 / Monday, March 26, 2012 / Rules and Regulations Date of issue: 08/18/2016 Version: 1.0 SECTION 1: Identification 1.1. Identification Product form : Substance Substance name : Iodomethane CAS No : 74-88-4 Product code : 1100-H-01 Formula : CH3I Synonyms : Methyl iodide Other means of identification : MFCD00001073 1.2. Relevant identified uses of the substance or mixture and uses advised against Use of the substance/mixture : Laboratory chemicals Manufacture of substances Scientific research and development 1.3. Details of the supplier of the safety data sheet SynQuest Laboratories, Inc. P.O. Box 309 Alachua, FL 32615 - United States of America T (386) 462-0788 - F (386) 462-7097 [email protected] - www.synquestlabs.com 1.4. Emergency telephone number Emergency number : (844) 523-4086 (3E Company - Account 10069) SECTION 2: Hazard(s) identification 2.1. Classification of the substance or mixture Classification (GHS-US) Acute Tox. 3 (Oral) H301 - Toxic if swallowed Acute Tox. 3 (Dermal) H311 - Toxic in contact with skin Acute Tox. 2 (Inhalation) H330 - Fatal if inhaled Acute Tox. 3 (Inhalation:vapour) H331 - Toxic if inhaled Skin Irrit. 2 H315 - Causes skin irritation Eye Dam. 1 H318 - Causes serious eye damage Resp. Sens. 1 H334 - May cause allergy or asthma symptoms or breathing difficulties if inhaled Skin Sens. 1 H317 - May cause an allergic skin reaction Carc. 2 H351 - Suspected of causing cancer STOT SE 3 H335 - May cause respiratory irritation -

METHYL IODIDE 1. Exposure Data
METHYL IODIDE Data were last reviewed in IARC (1986) and the compound was classified in IARC Monographs Supplement 7 (1987). 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Nomenclature Chem. Abstr. Serv. Reg. No.: 74-88-4 Chem. Abstr. Name: Iodomethane IUPAC Systematic Name: Iodomethane 1.1.2 Structural and molecular formulae and relative molecular mass H H C I H CH3I Relative molecular mass: 141.94 1.1.3 Chemical and physical properties of the pure substance (a) Description: Colourless transparent liquid, with a sweet ethereal odour (American Conference of Governmental Industrial Hygienists, 1992; Budavari, 1996) (b) Boiling-point: 42.5°C (Lide, 1997) (c) Melting-point: –66.4°C (Lide, 1997) (d) Solubility: Slightly soluble in water (14 g/L at 20°C); soluble in acetone; miscible with diethyl ether and ethanol (Budavari, 1996; Verschueren, 1996; Lide, 1997) (e) Vapour pressure: 53 kPa at 25.3°C; relative vapour density (air = 1), 4.9 (Ver- schueren, 1996) ( f ) Octanol/water partition coefficient (P): log P, 1.51 (Hansch et al., 1995) (g) Conversion factor: mg/m3 = 5.81 × ppm –1503– 1504 IARC MONOGRAPHS VOLUME 71 1.2 Production and use No information on the global production of methyl iodide was available to the Working Group. Production in the United States in 1983 was about 50 tonnes (IARC, 1986). Because of its high refractive index, methyl iodide is used in microscopy. It is also used as an embedding material for examining diatoms, in testing for pyridine, as a methy- lating agent in pharmaceutical (e.g., quaternary ammonium compounds) and chemical synthesis, as a light-sensitive etching agent for electronic circuits, and as a component in fire extinguishers (IARC, 1986; American Conference of Governmental Industrial Hygienists, 1992; Budavari, 1996). -

Synthesis of Bromochloromethane Using Phase Transfer Catalysis
1 SYNTHESIS OF BROMOCHLOROMETHANE USING PHASE TRANSFER CATALYSIS By LANCELOT LUCRETIUS BROOKS Baccalaureus Scientiae Honores-Chemistry, Nelson Mandela Metropolitan University A dissertation submitted in fulfillment of the requirements for the Masters Degree in Chemistry In the Faculty of Science at the NELSON MANDELA METROPOLITAN UNIVERSITY Nov. 2011 Promoter : Dr G. Dugmore Co-Promoter : Prof B. Zeelie 2 DECLARATION I, Lancelot Brooks, hereby declare that the above-mentioned treatise is my own work and that it has not previously been submitted for assessment to another University, or for another qualification. ……………………………….. ……………………….. Mr. L.L. Brooks Date 3 ACKNOWLEDGEMENTS To my promoters Dr. Gary Dugmore, and Prof. Ben Zeelie for their invaluable input, help and guidance. To NRF and NMMU for financial assistance To my parents and brothers for their love and support To Peter, Batsho, Unati, and friends in NMMU chemistry research laboratory, thank you guys. To my dearest fiancée, Natasha, a very special thank you for always being there and supporting me. Love you angel. “And we know that all things work together for good to those who love God, to those who are called according to His purpose” -Romans 8:28. 4 TABLE OF CONTENTS DECLARATION……………………………………………………………………. 2 ACKNOWLEDGEMENTS……………………………………………………………. 3 TABLE OF CONTENTS………………………………………………………………. 4 LIST OF FIGURES…………………………………………………………………….. 8 LIST OF TABLES……………………………………………………………………… 9 LIST OF EQUATIONS………………………………………………………………… 11 SUMMARY……………………………………………………………………………… 12 CHAPTER 1…………………………………………………………………………….. 14 INTRODUCTION………………………………………………………………………. 14 1.1. Technology of leather production……………………………………………….. 14 1.2. Synthesis of TCMTB……………………………………………………………… 17 1.3. Bromine……………………………………………………………………………. 20 1.3.1. Overview……………………………………………………………. 20 1.3.2. Applications of bromine compounds…………..…………………. 22 1.3.2.1. Photography……………………………………………… 22 1.3.2.2. -

Supplement to the Role of Internal Standards and Their Interaction With
Supplement to: Hiatt, M., “The Role of Internal Standards and their Interaction with Soils Impact Accuracy of Volatile Organics Determinations,” Int. J. of Environ. Anal. Chem., 2010 90:8 591-604. Please note that this is a non-copyrighted web version of supplemental information belonging to the above-cited journal article. There are formatting and page-numbering differences between this web version and the actual published version. There are also minor content differences. This version can be accessed at: http://www.epa.gov/nerlesd1/chemistry/vacuum/reference/pubs.htm This publication is also available from Informaworld: http://www.informaworld.com/smpp/content~db=all~content=a921288588 Web version, 27 April 2010 1 Supplement to: The Role of Internal Standards and their Interaction with Soils Impact Accuracy of Volatile Organics Determinations Michael H. Hiatt U.S. Environmental Protection Agency, National Exposure Research Laboratory Environmental Sciences Division. P.O. Box 93478, Las Vegas, Nevada 89193-3478 Phone: 702 798 2381. Fax: 702 798 2142. E-mail: [email protected]. Supplementary Information Data that was too detailed for “The Role of Internal Standards and their Interaction with Soils Impact Accuracy of Volatile Organics Determinations” is presented as supplemental information. This information includes each analyte by matrix and includes the accuracy evaluation, analyte results impacted by criteria, and results from varying spike equilibration times. The list of internal standards and their chemical properties are presented in Table S1 while the surrogates are listed in Table S2. Tables of results by analyte are included as Tables S3-8. These tables include the data by matrix and combined. -

Aldrich Organometallic, Inorganic, Silanes, Boranes, and Deuterated Compounds
Aldrich Organometallic, Inorganic, Silanes, Boranes, and Deuterated Compounds Library Listing – 1,523 spectra Subset of Aldrich FT-IR Library related to organometallic, inorganic, boron and deueterium compounds. The Aldrich Material-Specific FT-IR Library collection represents a wide variety of the Aldrich Handbook of Fine Chemicals' most common chemicals divided by similar functional groups. These spectra were assembled from the Aldrich Collections of FT-IR Spectra Editions I or II, and the data has been carefully examined and processed by Thermo Fisher Scientific. Aldrich Organometallic, Inorganic, Silanes, Boranes, and Deuterated Compounds Index Compound Name Index Compound Name 1066 ((R)-(+)-2,2'- 1193 (1,2- BIS(DIPHENYLPHOSPHINO)-1,1'- BIS(DIPHENYLPHOSPHINO)ETHAN BINAPH)(1,5-CYCLOOCTADIENE) E)TUNGSTEN TETRACARBONYL, 1068 ((R)-(+)-2,2'- 97% BIS(DIPHENYLPHOSPHINO)-1,1'- 1062 (1,3- BINAPHTHYL)PALLADIUM(II) CH BIS(DIPHENYLPHOSPHINO)PROPA 1067 ((S)-(-)-2,2'- NE)DICHLORONICKEL(II) BIS(DIPHENYLPHOSPHINO)-1,1'- 598 (1,3-DIOXAN-2- BINAPH)(1,5-CYCLOOCTADIENE) YLETHYNYL)TRIMETHYLSILANE, 1140 (+)-(S)-1-((R)-2- 96% (DIPHENYLPHOSPHINO)FERROCE 1063 (1,4- NYL)ETHYL METHYL ETHER, 98 BIS(DIPHENYLPHOSPHINO)BUTAN 1146 (+)-(S)-N,N-DIMETHYL-1-((R)-1',2- E)(1,5- BIS(DI- CYCLOOCTADIENE)RHODIUM(I) PHENYLPHOSPHINO)FERROCENY TET L)E 951 (1,5-CYCLOOCTADIENE)(2,4- 1142 (+)-(S)-N,N-DIMETHYL-1-((R)-2- PENTANEDIONATO)RHODIUM(I), (DIPHENYLPHOSPHINO)FERROCE 99% NYL)ETHYLAMIN 1033 (1,5- 407 (+)-3',5'-O-(1,1,3,3- CYCLOOCTADIENE)BIS(METHYLD TETRAISOPROPYL-1,3- IPHENYLPHOSPHINE)IRIDIUM(I) -

A High Volume Sampling System for Isotope Determination of Volatile Halocarbons and Hydrocarbons
Atmos. Meas. Tech., 4, 2073–2086, 2011 www.atmos-meas-tech.net/4/2073/2011/ Atmospheric doi:10.5194/amt-4-2073-2011 Measurement © Author(s) 2011. CC Attribution 3.0 License. Techniques A high volume sampling system for isotope determination of volatile halocarbons and hydrocarbons E. Bahlmann, I. Weinberg, R. Seifert, C. Tubbesing, and W. Michaelis Institute for Biogeochemistry and Marine Chemistry, Hamburg, Germany Received: 15 March 2011 – Published in Atmos. Meas. Tech. Discuss.: 8 April 2011 Revised: 31 August 2011 – Accepted: 7 September 2011 – Published: 4 October 2011 Abstract. The isotopic composition of volatile organic com- 1 Introduction pounds (VOCs) can provide valuable information on their sources and fate not deducible from mixing ratios alone. Compound specific isotope ratio mass spectrometry In particular the reported carbon stable isotope ratios of (CSIRMS) of non methane volatile organic compounds chloromethane and bromomethane from different sources (NMVOCs) emerged as a powerful tool to distinguish dif- 13 cover a δ C-range of almost 100 ‰ making isotope ra- ferent sources and to provide information on sinks (Rudolph tios a very promising tool for studying the biogeochemistry et al., 1997; Rudolph and Czuba, 2000; Tsunogai et al., of these compounds. So far, the determination of the iso- 1999; Bill et al., 2002; Thompson et al., 2002; Goldstein and topic composition of C1 and C2 halocarbons others than Shaw, 2003 and references therein; Archbold et al., 2005; chloromethane is hampered by their low mixing ratios. Redeker -

Methyl Iodide (Iodomethane)
Methyl Iodide (Iodomethane) 74-88-4 Hazard Summary Methyl iodide is used as an intermediate in the manufacture of some pharmaceuticals and pesticides, in methylation processes, and in the field of microscopy. In humans, acute (short-term) exposure to methyl iodide by inhalation may depress the central nervous system (CNS), irritate the lungs and skin, and affect the kidneys. Massive acute inhalation exposure to methyl iodide has led to pulmonary edema. Acute inhalation exposure of humans to methyl iodide has resulted in nausea, vomiting, vertigo, ataxia, slurred speech, drowsiness, skin blistering, and eye irritation. Chronic (long-term) exposure of humans to methyl iodide by inhalation may affect the CNS and cause skin burns. EPA has not classified methyl iodide for potential carcinogenicity. Please Note: The main sources of information for this fact sheet are the International Agency for Research on Cancer (IARC) monographs on chemicals carcinogenic to humans (2) and the Hazardous Substances Data Bank (HSDB) (3), a database of summaries of peer-reviewed literature. Uses Methyl iodide is used as an intermediate in the manufacture of some pharmaceuticals and pesticides. It is also used in methylation processes and in the field of microscopy. (1,2,4) Proposed uses of methyl iodide are as a fire extinguisher and as an insecticidal fumigant. (5) Sources and Potential Exposure Individuals are most likely to be exposed to methyl iodide in the workplace. (1) Methyl iodide occurs naturally in the ocean as a product of marine algae. (2) Assessing Personal Exposure No information was located regarding the measurement of personal exposure to methyl iodide. -

Measurement of Formic and Acetic Acid in Air by Chemical Ionization Mass Spectroscopy: Airborne Method Development
University of Rhode Island DigitalCommons@URI Open Access Master's Theses 2015 Measurement of Formic and Acetic Acid in Air by Chemical Ionization Mass Spectroscopy: Airborne Method Development Victoria Treadaway University of Rhode Island, [email protected] Follow this and additional works at: https://digitalcommons.uri.edu/theses Recommended Citation Treadaway, Victoria, "Measurement of Formic and Acetic Acid in Air by Chemical Ionization Mass Spectroscopy: Airborne Method Development" (2015). Open Access Master's Theses. Paper 603. https://digitalcommons.uri.edu/theses/603 This Thesis is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Master's Theses by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. MEASUREMENT OF FORMIC AND ACETIC ACID IN AIR BY CHEMICAL IONIZATION MASS SPECTROSCOPY: AIRBORNE METHOD DEVELOPMENT BY VICTORIA TREADAWAY A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE IN OCEANOGRAPHY UNIVERSITY OF RHODE ISLAND 2015 MASTER OF SCIENCE THESIS OF VICTORIA TREADAWAY APPROVED: Thesis Committee: Major Professor Brian Heikes John Merrill James Smith Nasser H. Zawia DEAN OF THE GRADUATE SCHOOL UNIVERSITY OF RHODE ISLAND 2015 ABSTRACT The goals of this study were to determine whether formic and acetic acid could be quantified from the Deep Convective Clouds and Chemistry Experiment (DC3) through post-mission calibration and analysis and to optimize a reagent gas mix with CH3I, CO2, and O2 that allows quantitative cluster ion formation with hydroperoxides and organic acids suitable for use in future field measurements. -

Gas Phase Chemistry and Removal of CH3I During a Severe Accident
DK0100070 Nordisk kernesikkerhedsforskning Norraenar kjarnoryggisrannsoknir Pohjoismainenydinturvallisuustutkimus Nordiskkjernesikkerhetsforskning Nordisk karnsakerhetsforskning Nordic nuclear safety research NKS-25 ISBN 87-7893-076-6 Gas Phase Chemistry and Removal of CH I during a Severe Accident Anna Karhu VTT Energy, Finland 2/42 January 2001 Abstract The purpose of this literature review was to gather valuable information on the behavior of methyl iodide on the gas phase during a severe accident. The po- tential of transition metals, especially silver and copper, to remove organic io- dides from the gas streams was also studied. Transition metals are one of the most interesting groups in the contex of iodine mitigation. For example silver is known to react intensively with iodine compounds. Silver is also relatively inert material and it is thermally stable. Copper is known to react with some radioio- dine species. However, it is not reactive toward methyl iodide. In addition, it is oxidized to copper oxide under atmospheric conditions. This may limit the in- dustrial use of copper. Key words Methyl iodide, gas phase, severe accident mitigation NKS-25 ISBN 87-7893-076-6 Danka Services International, DSI, 2001 The report can be obtained from NKS Secretariat P.O. Box 30 DK-4000RoskiIde Denmark Phone +45 4677 4045 Fax +45 4677 4046 http://www.nks.org e-mail: [email protected] Gas Phase Chemistry and Removal of CH3I during a Severe Accident VTT Energy, Finland Anna Karhu Abstract The purpose of this literature review was to gather valuable information on the behavior of methyl iodide on the gas phase during a severe accident. -
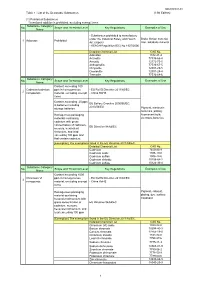
Table 1 : List of the Declarable Substances (1) Prohibited
DQ2000001-03 Table 1 : List of the Declarable Substances (13th Edition) (1) Prohibited Substances *Intentional addition is prohibited, excluding exempt items Substance Category/ No. Scope and Threshold Level Key Regulations Examples of Use Name - Substances prohibited to manufacture under the Industrial Safety and Health Brake friction material, 1 Asbestos Prohibited Act (Japan) filler, adiabatic material - REACH Regulation (EC) No 1907/2006 Detailed Chemical List CAS No. Asbestos 1332-21-4 Actinolite 77536-66-4 Amosite 12172-73-5 Anthophyllite 77536-67-5 Chrysotile 12001-29-5 Crocidolite 12001-28-4 Tremolite 77536-68-6 Substance Category/ No. Scope and Threshold Level Key Regulations Examples of Use Name Content exceeding 100 Cadmium/cadmium ppm in homogeneous - EU RoHS Directive 2011/65/EC 2 compounds material, excluding exempt - China RoHS items Content exceeding 20 ppm EU Battery Directive 2006/66/EC, in batteries including 2013/56/EU storage batteries Pigment, electronic materials, plating, Homogenous packaging fluorescent bulb, materials containing electrode,batteries cadmium with gross concentration of cadmium, EU Directive 94/62/EC mercury, hexavalent chromium, and lead exceeding 100 ppm and that contain cadmium [Exemption] The exemptions listed in the EU Directive 2011/65/EC. Detailed Chemical List CAS No. Cadmium 7440-43-9 Cadmium oxide 1306-19-0 Cadmium sulfide 1306-23-6 Cadmium chloride 10108-64-2 Cadmium sulfate 10124-36-4 Substance Category/ No. Scope and Threshold Level Key Regulations Examples of Use Name Content exceeding 1000 Chromium VI ppm in homogeneous - EU RoHS Directive 2011/65/EC 3 compounds material, excluding exempt - China RoHS items Homogenous packaging Pigment, catalyst, material containing plating, dye, surface hexavalent chromium with treatment gross concentration of EU Directive 94/62/EC cadmium, mercury, hexavalent chromium, and lead exceeding 100 ppm [Exemption] The exemptions listed in the EU Directive 2011/65/EC. -

SROC Annex V
Annex V Major Chemical Formulae and Nomenclature This annex presents the formulae and nomenclature for halogen-containing species and other species that are referred to in this report (Annex V.1). The nomenclature for refrigerants and refrigerant blends is given in Annex V.2. V.1 Substances by Groupings V.1.1 Halogen-Containing Species V.1.1.1 Inorganic Halogen-Containing Species Atomic chlorine Cl Atomic bromine Br Molecular chlorine Cl2 Molecular bromine Br2 Chlorine monoxide ClO Bromine monoxide BrO Chlorine radicals ClOx Bromine radicals BrOx Chloroperoxy radical ClOO Bromine nitrate BrONO2, BrNO3 Dichlorine peroxide (ClO dimer) (ClO)2, Cl2O2 Potassium bromide KBr Hydrogen chloride (Hydrochloric acid) HCl Inorganic chlorine Cly Antimony pentachloride SbCl5 Atomic fluorine F Molecular fluorine F2 Atomic iodine I Hydrogen fluoride (Hydrofluoric acid) HF Molecular iodine I2 Sulphur hexafluoride SF6 Nitrogen trifluoride NF3 IPCC Boek (dik).indb 467 15-08-2005 10:57:13 468 IPCC/TEAP Special Report: Safeguarding the Ozone Layer and the Global Climate System V.1.1.2 Halocarbons For each halocarbon the following information is given in columns: • Chemical compound [Number of isomers]1 (or common name) • Chemical formula • CAS number2 • Chemical name (or alternative name) V.1.1.2.1 Chlorofluorocarbons (CFCs) CFC-11 CCl3F 75-69-4 Trichlorofluoromethane CFC-12 CCl2F2 75-71-8 Dichlorodifluoromethane CFC-13 CClF3 75-72-9 Chlorotrifluoromethane CFC-113 [2] C2Cl3F3 Trichlorotrifluoroethane CCl FCClF 76-13-1 CFC-113 2 2 1,1,2-Trichloro-1,2,2-trifluoroethane