Geographical Structuring in the Mtdna of Italians
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Corso Di Formazione Per Operatore Socio Sanitario
UNIONE EUROPEA PROVINCIA DI NUORO Fondo Sociale Europeo Assessorato del Lavoro, Formazione Professionale, Cooperazione e Sicurezza sociale SETTORE LAVORO, FORMAZIONE PROFESSIONALE E POLITICHE SOCIALI AVVISO PUBBLICO PER L’ATTUAZIONE DEL PIANO PROVINCIALE DI FORMAZIONE PROFESSIONALE ANNUALITÀ 2011-2012 CIG. 4810202044 CORSO DI FORMAZIONE PER OPERATORE SOCIO SANITARIO GRADUATORIA RESIDENTI IN PROVINCIA DI NUORO IN ORDINE DI PUNTEGGIO Sono evidenziati in verde gli ammessi alla prova orale Prova 1 Prova 2 Titoli Titoli Valutazione Test Posizione Cognome Nome Data di nascita Luogo residenza Prov residenza Esperienza disoccupazione TOTALE competenze psicoattitudinale (MAX 17 Punti) (MAX 8 punti) (MAX 25 punti) (MAX 30 punti) 1 SANNA MARIA FRANCESCA 08/11/1972 MEANA SARDO NUORO 4,8 4 20 28,23 57,03 2 SALIS PIETRINA 22/03/1978 OLIENA NUORO 5 8 21 22,64 56,64 3 TIDU BARBARA 23/09/1971 TETI NUORO 12,2 8 23 12,33 55,53 4 CURRELI GIUSEPPINA 16/11/1977 ARITZO NUORO 13 1,5 25 15,58 55,08 5 SEDDA MARTA 13/08/1984 OLLOLAI NUORO 12 1,5 25 15,49 53,99 6 CONGIU GIOVANNA MARIA 08/12/1973 SILANUS NUORO 4,8 8 25 15,80 53,60 7 LICHERI BARBARA 29/12/1977 OLZAI NUORO 9 0 25 19,31 53,31 8 SECHI JENNIFER MARIA LUISA 16/11/1980 BORORE NUORO 7,8 8 25 12,34 53,14 9 DELIGIA SALVATORANGELA 29/06/1981 BORORE NUORO 12,4 1,5 25 14,18 53,08 10 MORO ANTONIETTA 30/09/1966 OLZAI NUORO 10 8 23 12,01 53,01 11 MELONI RITA 05/09/1966 ORTUERI NUORO 15,2 8 15 14,68 52,88 12 CARBONI FRANCESCA 31/10/1969 TONARA NUORO 17 8 12 15,64 52,64 13 TALLORU SERGIO MAURO 26/05/1985 TONARA NUORO -

Veneto Main Cities and Key Economic Sectors
VENETO MAIN CITIES AND KEY ECONOMIC SECTORS OVERVIEW – Veneto Region Area: 18.345,35 km2 Corporate taxes: Provinces: Padova, Rovigo, Vicenza, 0-15.000 € 23% Verona, Treviso, Venezia, Belluno 15.001-28.000 € 27% Municipalities: 94 28.001-55.000 € 38% 55.001-75.000 € 41% Population: 4.907.529 75.001 € > … € 43% Capital of the region: Venice Additional regional GDP: Taxable income < 45.000 € 0,9% Language: Italian Taxable icome > 300.000 € 0,9 % GDP (2017): 162,5 billion Euro + 3% solidarity contribution GDP per capita (2017): 33.122 Euro GDP annual growth (2017): +2.3% Source: statistica.regione.veneto.it OVERVIEW – Venetian provinces and main cities of Veneto Region Padova: 936 274 inhabitants Venezia Today magazine…. “The image of the productive and entrepreneurial Northeast also owes much to Verona: 921 557 inhabitants Treviso. In just a few years an area with an almost agricultural economy, a sector Treviso: 885 972 inhabitants still very flourishing and specialized (as confirmed by the vineyards of Conegliano and Valdobbiadene, with the renowned Prosecco Superiore DOCG and Vicenza: 865 082 inhabitants radicchio Treviso), has become one of the engines of the Italian economy, in Venezia: 854 275 inhabitants particular in the mechanical sectors, metalworking, textiles, food and clothing, in which it has been able to establish itself worldwide with some prestigious brands Rovigo: 238 558 inhabitants such as Benetton. The main industrial districts concern furniture, stainless steel products, footwear and sportswear.” Belluno: 205 -

Mattu Antonina
F ORMATO EUROPEO PER IL CURRICULUM VITAE INFORMAZIONI PERSONALI Nome MATTU ANTONINA Qualifica SEGRETARIO GENERALE Amministrazione COMUNE DI LANUSEI (NU) Telefono 0782/473130 E-mail [email protected] - ([email protected]) Nazionalità Italiana Data di nascita OMISSIS … ESPERIENZA LAVORATIVA DA DICEMBRE 2016 AD OGGI SEGRETARIO GENERALE DELLA CONVENZIONE TRA IL COMUNE DI LANUSEI E I COMUNI DI OLZAI E OLLOLAI OLLOLAI (NU) • INCARICO DI PRESIDENTE NUCLEO VALUTAZIONE • INCARICO DI PRESIDENTE DELLA DELEGAZIONE TRATTANTE DI PARTE PUBBLICA • INCARICO DI PRESIDENTE DELLA COMMISSIONE PER I PROCEDIMENTI DISCIPLINARI • SOSTITUZIONE RESPONSABILI PP.OO.. • RESPONSABILE ANTICORRUZIONE E TRASPARENZA DA OTTOBRE 2005 AD AGOSTO SEGRETARIO GENERALE – SEDE TITOLARE COMUNE SINISCOLA .(NU) 2016 • NOMINA DI DIRETTORE (FINO AL MAGGIO 2016) • INCARICO DI PRESIDENTE NUCLEO VALUTAZIONE , DELLA DELEGAZIONE TRATTANTE • RESPONSABILE UFFICIO PER I PROCEDIMENTI DISCIPLINARI • SOSTITUZIONE RESPONSABILI LE PP.OO. IN CASO DI ASSENZA. • RESPONSABILE ANTICORRUZIONE (DAL 24/01/2014) • SUPPLENZE A SCAVALCO PRESSO I COMUNI DI BITTI, LOCULI, OLZAI E OLLOLAI (NU) DAL GENNAIO 2001 FINO A TITOLARE SEGRETERIA CONSORZIATA DEI COMUNI DI TONARA DESULO (NU) SETTEMBRE 2005 SUPPLENZE A SCAVALCO PRESSO I COMUNI DI ARITZO, BELVÌ, GADONI(NU) DAL MAGGIO 1997 FINO AL GENNAIO SEGRETARIO COMUNALE – SEDE TITOLARE COMUNE TONARA.(NU) 2001 • INCARICO DI PRESIDENTE NUCLEO VALUTAZIONE, • PRESIDENTE DELLA DELEGAZIONE TRATTANTE • RESP SERVIZIO AMM.VO E SOSTITUZIONE RESPONSABILI DI SERVIZIO IN CASO DI ASSENZA. • SUPPLENZE A SCAVALCO PRESSO I COMUNI DI ARITZO, BELVÌ E DESULO(NU) DAL FEBBRAIO AL MAGGIO 1997 SEGRETARIO COMUNALE – SEDE TITOLARE COMUNE DI OVODDA (NU) DAL 1990 AL 1996 SEGRETARIO COMUNALE – SEDE TITOLARE COMUNE DI TIANA.(NU) SUPPLENZE A SCAVALCO PRESSO I COMUNI DI ARITZO, AUSTIS, BELVÌ, GAVOI, MAMOIADA, TONARA E TETI. -

A History of Italian Literature Should Follow and Should Precede Other and Parallel Histories
I. i III 2.3 CORNELL UNIVERSITY LIBRARY C U rar,y Ubrary PQ4038 G°2l"l 8t8a iterature 1lwBiiMiiiiiiiifiiliiii ! 3 1924 oim 030 978 245 Date Due M#£ (£i* The original of this book is in the Cornell University Library. There are no known copyright restrictions in the United States on the use of the text. http://www.archive.org/details/cu31924030978245 Short Histories of the Literatures of the World: IV. Edited by Edmund Gosse Short Histories of the Literatures of the World Edited by EDMUND GOSSE Large Crown 8vOj cloth, 6s. each Volume ANCIENT GREEK LITERATURE By Prof. Gilbert Murray, M.A. FRENCH LITERATURE By Prof. Edward Dowden, D.C.L., LL.D. MODERN ENGLISH LITERATURE By the Editor ITALIAN LITERATURE By Richard Garnett, C.B., LL.D. SPANISH LITERATURE By J. Fitzmaurice-Kelly [Shortly JAPANESE LITERATURE By William George Aston, C.M.G. [Shortly MODERN SCANDINAVIAN LITERATURE By George Brandes SANSKRIT LITERATURE By Prof. A. A. Macdonell. HUNGARIAN LITERATURE By Dr. Zoltan Beothy AMERICAN LITERATURE By Professor Moses Coit Tyler GERMAN LITERATURE By Dr. C. H. Herford LATIN LITERATURE By Dr. A. W. Verrall Other volumes will follow LONDON: WILLIAM HEINEMANN \AU rights reserved] A .History of ITALIAN LITERATURE RICHARD GARNETT, C.B., LL.D. Xon&on WILLIAM HEINEMANN MDCCCXCVIII v y. 1 1- fc V- < V ml' 1 , x.?*a»/? Printed by Ballantyne, Hanson &* Co. At the Ballantyne Press *. # / ' ri PREFACE "I think," says Jowett, writing to John Addington Symonds (August 4, 1890), "that you are happy in having unlocked so much of Italian literature, certainly the greatest in the world after Greek, Latin, English. -

Pier Virgilio Arrigoni the Discovery of the Sardinian Flora
Pier Virgilio Arrigoni The discovery of the Sardinian Flora (XVIII-XIX Centuries) Abstract Arrigoni, P. V.: The discovery of the Sardinian Flora (XVIII-XIX Centuries). — Bocconea 19: 7-31. 2006. — ISSN 1120-4060. The history of the floristic exploration of Sardinia mainly centres round the works of G.G. Moris, who in the first half of the XIX century described most of the floristic patrimony of the island. But it is important to know the steps he took in his census, the areas he explored, his publications, motivations and conditions under which he wrote the "Stirpium sardoarum elenchus" and the three volumes of "Flora sardoa", a work moreover which he left incomplete. Merit is due to Moris for bringing the attention of many collectors, florists and taxonomists to the Flora of the Island, individuals who in his foot-steps helped to complete and update the floristic inventory of the island. Research into the history of our knowledge of the Sardinian Flora relies heavily on the analysis of botanical publications, but many other sources (non- botanical texts, chronicles of the period, correspondence) also furnish important information. Finally, the names, dates and collection localities indicated on the specimens preserved in the most important herbaria were fundamental in reconstructing the itineraries of the sites Moris visited. All these sources allowed us to clarify several aspects of the expeditions, floristic col- lections and results of his studies. The "discovery phase" of Sardinian Flora can be considered over by the end of the XIX century with the publication of the "Compendium" by Barbey (1884-1885) and "Flora d'Italia" by Fiori & Paoletti (1896-1908). -

Graduatoria Idonei - Eta' Superiore a 25 Anni
Unione Europea Repubblica Italiana REGIONE AUTONOMA DE SARDIGNA Fondo Sociale Europeo REGIONE AUTONOMA DELLA SARDEGNA Assessoradu de su Traballu, Formatzione Professionale, Cooperatzione e Segurantzia Sotziale Assessorato del Lavoro Formazione Professionale, Cooperazione e Sicurezza Sociale CORSO DI QUALIFICA PER OPERATORE SOCIO-SANITARIO. - DISOCCUPATI/INOCCUPATI- LOTTO N.8 AZIONE "A" PROV. NUORO Selezione Candidati GRADUATORIA IDONEI - ETA' SUPERIORE A 25 ANNI AGENZIA FORMATIVA EVOLVERE POSIZIONE PUNTEGGIO NOMINATIVO DATA DI NASCITA LUOGO DI NASCITA COMUNE DI RESIDENZA NOTE GRADUATORIA PROVINCIA DI COMPLESSIVO RESIDENZA (sigla) 1 Sale Loretina 10/10/56 Mamoiada Mamoiada NU 75 2 Ganga Graziano 18/12/57 Nuoro Nuoro NU 70 3 Fele Giovanna Maria 07/09/60 Oliena Oliena NU 70 4 Gioi Anna 27/02/64 Desulo Desulo NU 70 5 Sanna Adriana 23/10/68 Solarussa Ovodda NU 70 6 Sotgiu Antonella 31/05/70 Sorgono Belvì NU 70 7 Zedda Graziella 21/08/71 Tiana Tiana NU 70 8 Picca Enrica 30/09/75 Nuoro Oliena NU 70 9 Moi Milvia 10/01/68 Urzulei Nuoro NU 65 10 Mureddu Tiziana 16/07/74 Austis Austis NU 65 11 Nuvoli Elisa 10/04/76 Nuoro Ollollai NU 65 12 Zoroddu Tonina 24/07/84 Nuoro Ottana NU 65 13 Mura Valentina 11/08/84 Nuoro Silanus NU 65 14 Morittu Giovanna 30/12/84 Nuoro Orotelli NU 65 15 Nieddu Caterina 21/08/72 Dorgali Dorgali NU 61,35 16 Sale Maria Grazia 12/02/81 Nuoro Fonni NU 60,6 17 Manca Grazia 23/05/60 Nuoro Nuoro NU 60 18 Curreli Rosalba 01/03/63 Ovodda Ovodda NU 60 La Croce Battistina 19 16/07/64 Tonara Tonara NU 60 Carmen 20 Carta Anna Rita 14/06/66 -

Representations of Italian Americans in the Early Gilded Age
Differentia: Review of Italian Thought Number 6 Combined Issue 6-7 Spring/Autumn Article 7 1994 From Italophilia to Italophobia: Representations of Italian Americans in the Early Gilded Age John Paul Russo Follow this and additional works at: https://commons.library.stonybrook.edu/differentia Recommended Citation Russo, John Paul (1994) "From Italophilia to Italophobia: Representations of Italian Americans in the Early Gilded Age," Differentia: Review of Italian Thought: Vol. 6 , Article 7. Available at: https://commons.library.stonybrook.edu/differentia/vol6/iss1/7 This document is brought to you for free and open access by Academic Commons. It has been accepted for inclusion in Differentia: Review of Italian Thought by an authorized editor of Academic Commons. For more information, please contact [email protected], [email protected]. From ltalophilia to ltalophobia: Representations of Italian Americans in the Early Gilded Age John Paul Russo "Never before or since has American writing been so absorbed with the Italian as it is during the Gilded Age," writes Richard Brodhead. 1 The larger part of this American fascination expressed the desire for high culture and gentility, or what Brodhead calls the "aesthetic-touristic" attitude towards Italy; it resulted in a flood of travelogues, guidebooks, antiquarian stud ies, historical novels and poems, peaking at the turn of the centu ry and declining sharply after World War I. America's golden age of travel writing lasted from 1880 to 1914, and for many Americans the richest treasure of all was Italy. This essay, however, focuses upon Brodhead's other catego ry, the Italian immigrant as "alien-intruder": travel writing's gold en age corresponded exactly with the period of greatest Italian immigration to the United States. -

Italian Immigrants and Italy: an Introduction to the Multi-Media Package on Italy
DOCUMENT RESUME ED 067 332 SO 004 339 AUTHOR Witzel, Anne TITLE Italian Immigrants and Italy: An Introduction to the Multi-Media Package on Italy. INSTITUTION Toronto Board of Education (Ontario). Research Dept. PUB DATE May 69 NOTE 16p. EDRS PRICE MF-$0.65 HC -$ 3.29 DESCRIPTORS Annotated Bibliographies; *Cultural Background; Elementary Education; *European History; Geography; History; *Immigrants; *Italian Literature; Resource Guides; Secondary Education IDENTIFIERS *Italy ABSTRACT The largest group of non-English speaking immigrants who come to Canada are Italians, the vast majority of whom are from Southern Italy. This paper furnishes information on their cultural background and lists multi-media resources to introduce teachers to Italian society so that educators may better understand their students. Immigrant children are faced with choosing between two conflicting life styles -- the values of Canadian society and family values and customs. When teachers are aware of the problem they can cushion the culture shock for students and guide them througha transitional period. The paper deals with history, geography, and climate, explaining and suggesting some ideas on why Southern Italy differs from Northern and Central Italy. Cultural differencescan be traced not only to the above factors, but also to ethnic roots and the "culture of poverty" -- attitudes of the poor which create a mentality that perpetuates living at a subsistence level. The low status of women as it affects society is discussed, since the family is seen as a society in microcosm. The last portion of the paper presents primary sources, annotated bibliographies, and audio-visual materials. A related document is SO 004 351. -
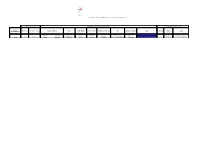
1 179 20171130160418.Pdf
Il Responsabile dell'Unità di Progetto per l'eradicazione della peste suina africana Areale di caccia Referente e delegato gruppo di caccia Locale di cui all'art. 5.1.2 lett. b. cod id. Provincia Comuni di caccia Cognome Nome Ruolo n. Aut. Reg.le n. Porto armi Residenza (Comune) Via Telefono cellulare mail Provincia Comune Località Autorizzazione PUDDU STEFANO REFERENTE 430641 894354-N ESTERZILI BORSELLINO, 6 3420968298 [email protected] Cagliari Esterzili CA ESTERZILI VIA SANTA MARIA,22 Z108/45 DESSI' ANDREA DELEGATO 430634 892262-N ESTERZILI Vico E. D'ARBOREA 3472822045 [email protected] Il Responsabile dell'Unità di Progetto per l'eradicazione della peste suina africana cod id. Compagnia Provincia Comuni di caccia Sostituto referente Referente Responsabile n. Aut. Reg.le n. Porto armi Residenza (Comune) Via Telefono cellulare mail Provincia Comune Località Latitudine WGS84 Longitudine WGS84 Cod.Autorizzazione Z104/115 MARONGIU LANUSEI OG LANUSEI-ARZANA-GAIRO-VILLAGRANDE-SEUI-TALANA-ORGOSOLO FRANCESCHI ROBRTO MARONGIU ENZO TOMASO 482928-N 407108 LANUSEI SPANO 87 3479447611 [email protected] OG LANUSEI G.GALILEI 39,5243 9,324613 Z104/116 OG CARDEDU LORRAI SALVATORE DEMURTAS MARIO BRUNO 423645 658957-N CARDEDU CAGLIARI, 1 3498561632 OG CARDEDU BACCU ORRODAS Z104/117 SQUADRA DE MESUDI OG GAIRO BRENDAS ROBERTO ZEDDA ROBERTO 436453-N 891947-N GAIRO VIA MANNU 18 3298839842 OG GAIRO COLOMBO Z104/118 OG JERZU SALIS ANDREA CASULA ANTONIO 414647-N 656939-N JERZU A.BUSINCO 3389636052 OG JERZU A.BUSINCO Z104/119 SANT'ANTONIO OG JERZU MURA MARIO SERRA SALVATORE 384098 891248-N JERZU E.FERMI 26 3335477257 [email protected] OG JERZU E. -

The Ancient People of Italy Before the Rise of Rome, Italy Was a Patchwork
The Ancient People of Italy Before the rise of Rome, Italy was a patchwork of different cultures. Eventually they were all subsumed into Roman culture, but the cultural uniformity of Roman Italy erased what had once been a vast array of different peoples, cultures, languages, and civilizations. All these cultures existed before the Roman conquest of the Italian Peninsula, and unfortunately we know little about any of them before they caught the attention of Greek and Roman historians. Aside from a few inscriptions, most of what we know about the native people of Italy comes from Greek and Roman sources. Still, this information, combined with archaeological and linguistic information, gives us some idea about the peoples that once populated the Italian Peninsula. Italy was not isolated from the outside world, and neighboring people had much impact on its population. There were several foreign invasions of Italy during the period leading up to the Roman conquest that had important effects on the people of Italy. First there was the invasion of Alexander I of Epirus in 334 BC, which was followed by that of Pyrrhus of Epirus in 280 BC. Hannibal of Carthage invaded Italy during the Second Punic War (218–203 BC) with the express purpose of convincing Rome’s allies to abandon her. After the war, Rome rearranged its relations with many of the native people of Italy, much influenced by which peoples had remained loyal and which had supported their Carthaginian enemies. The sides different peoples took in these wars had major impacts on their destinies. In 91 BC, many of the peoples of Italy rebelled against Rome in the Social War. -

Read Kansas!Kansas! by the Kansas State Historical Society
rki8_italian.qxp 4/22/08 2:51 PM Page 1 YOUR KANSAS STORIES OUR HISTORICAL I-8 HISTORY SOCIETY ReadRead Kansas!Kansas! By the Kansas State Historical Society Italians in Kansas: The Story of Pittsburg The history of southeast Kansas is connected to coal mining. In 1874 the first underground coal mine opened. Many immigrants, including Italians, came to Kansas to work in the mines. Italy is a country in Europe. Why did they leave Italy? Italy had too many people. It was too crowded. The wages were low and the taxes were high. Many Italians wanted better economic opportunities. Most of the Italians who came to the United States were from rural areas. They had limited education. They hoped to find work in the growing industries of America. Men immigrated more often than women. Some planned to come, earn money, and return to Italy. Others sent money home to pay for their families to join them in America. Why did they come to Kansas? Pittsburg is a city in Crawford County. Many coal mines surround the city. In 1878 miners in southeast Kansas went on strike. By refusing to work, they hoped to get the mine owners’ attention. They wanted better pay and better working conditions. The mine owners looked for other workers. Workers from northern Italy were recruited. New workers poured into Kansas to work in the mines. Within fifteen years, 1,200 Italians had moved into the areas around Pittsburg. In another 20 years, that number tripled. What did they find in Kansas? Coal mines attracted most Italians to Kansas. -

The Roman Army's Emergence from Its Italian Origins
CORE Metadata, citation and similar papers at core.ac.uk Provided by Carolina Digital Repository THE ROMAN ARMY’S EMERGENCE FROM ITS ITALIAN ORIGINS Patrick Alan Kent A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of History. Chapel Hill 2012 Approved by: Richard Talbert Nathan Rosenstein Daniel Gargola Fred Naiden Wayne Lee ABSTRACT PATRICK ALAN KENT: The Roman Army’s Emergence from its Italian Origins (Under the direction of Prof. Richard Talbert) Roman armies in the 4 th century and earlier resembled other Italian armies of the day. By using what limited sources are available concerning early Italian warfare, it is possible to reinterpret the history of the Republic through the changing relationship of the Romans and their Italian allies. An important aspect of early Italian warfare was military cooperation, facilitated by overlapping bonds of formal and informal relationships between communities and individuals. However, there was little in the way of organized allied contingents. Over the 3 rd century and culminating in the Second Punic War, the Romans organized their Italian allies into large conglomerate units that were placed under Roman officers. At the same time, the Romans generally took more direct control of the military resources of their allies as idea of military obligation developed. The integration and subordination of the Italians under increasing Roman domination fundamentally altered their relationships. In the 2 nd century the result was a growing feeling of discontent among the Italians with their position.