Xenical, INN-Orlistat
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Pharmacology of Amiodarone and Digoxin As Antiarrhythmic Agents
Part I Anaesthesia Refresher Course – 2017 University of Cape Town The Pharmacology of Amiodarone and Digoxin as Antiarrhythmic Agents Dr Adri Vorster UCT Department of Anaesthesia & Perioperative Medicine The heart contains pacemaker, conduction and contractile tissue. Cardiac arrhythmias are caused by either enhancement or depression of cardiac action potential generation by pacemaker cells, or by abnormal conduction of the action potential. The pharmacological treatment of arrhythmias aims to achieve restoration of a normal rhythm and rate. The resting membrane potential of myocytes is around -90 mV, with the inside of the membrane more negative than the outside. The main extracellular ions are Na+ and Cl−, with K+ the main intracellular ion. The cardiac action potential involves a change in voltage across the cell membrane of myocytes, caused by movement of charged ions across the membrane. This voltage change is triggered by pacemaker cells. The action potential is divided into 5 phases (figure 1). Phase 0: Rapid depolarisation Duration < 2ms Threshold potential must be reached (-70 mV) for propagation to occur Rapid positive charge achieved as a result of increased Na+ conductance through voltage-gated Na+ channels in the cell membrane Phase 1: Partial repolarisation Closure of Na+ channels K+ channels open and close, resulting in brief outflow of K+ and a more negative membrane potential Phase 2: Plateau Duration up to 150 ms Absolute refractory period – prevents further depolarisation and myocardial tetany Result of Ca++ influx -

Dabigatran Amoxicillin Clavulanate IV Treatment in the Community
BEST PRACTICE 38 SEPTEMBER 2011 Dabigatran Amoxicillin clavulanate bpac nz IV treatment in the community better medicin e Editor-in-chief We would like to acknowledge the following people for Professor Murray Tilyard their guidance and expertise in developing this edition: Professor Carl Burgess, Wellington Editor Dr Gerry Devlin, Hamilton Rebecca Harris Dr John Fink, Christchurch Dr Lisa Houghton, Dunedin Programme Development Dr Rosemary Ikram, Christchurch Mark Caswell Dr Sisira Jayathissa, Wellington Rachael Clarke Kate Laidlow, Rotorua Peter Ellison Dr Hywel Lloyd, GP Reviewer, Dunedin Julie Knight Associate Professor Stewart Mann, Wellington Noni Richards Dr Richard Medlicott, Wellington Dr AnneMarie Tangney Dr Alan Panting, Nelson Dr Sharyn Willis Dr Helen Patterson, Dunedin Dave Woods David Rankin, Wellington Report Development Dr Ralph Stewart, Auckland Justine Broadley Dr Neil Whittaker, GP Reviewer, Nelson Tim Powell Dr Howard Wilson, Akaroa Design Michael Crawford Best Practice Journal (BPJ) ISSN 1177-5645 Web BPJ, Issue 38, September 2011 Gordon Smith BPJ is published and owned by bpacnz Ltd Management and Administration Level 8, 10 George Street, Dunedin, New Zealand. Jaala Baldwin Bpacnz Ltd is an independent organisation that promotes health Kaye Baldwin care interventions which meet patients’ needs and are evidence Tony Fraser based, cost effective and suitable for the New Zealand context. Kyla Letman We develop and distribute evidence based resources which describe, facilitate and help overcome the barriers to best Clinical Advisory Group practice. Clive Cannons nz Michele Cray Bpac Ltd is currently funded through contracts with PHARMAC and DHBNZ. Margaret Gibbs nz Dr Rosemary Ikram Bpac Ltd has five shareholders: Procare Health, South Link Health, General Practice NZ, the University of Otago and Pegasus Dr Cam Kyle Health. -

Lifestyle Drugs” for Men and Women
Development of “Lifestyle Drugs” for Men and Women Armin Schultz CRS - Clinical Research Services Mannheim GmbH AGAH Annual Meeting 2012, Leipzig, March 01 - 02 Lifestyle drugs Smart drugs, Quality-of-life drugs, Vanity drugs etc. Lifestyle? Lifestyle-Drugs? Active development? Discovery by chance? AGAH Annual Meeting 2012, Leipzig, March 01 - 02 Lifestyle A lifestyle is a characteristic bundle of behaviors that makes sense to both others and oneself in a given time and place, including social relations, consumption, entertainment, and dress. The behaviors and practices within lifestyles are a mixture of habits, conventional ways of doing things, and reasoned actions „Ein Lebensstil ist [...] der regelmäßig wiederkehrende Gesamtzusammenhang der Verhaltensweisen, Interaktionen, Meinungen, Wissensbestände und bewertenden Einstellungen eines Menschen“ (Hradil 2005: 46) Different definitions in social sciences, philosophy, psychology or medicine AGAH Annual Meeting 2012, Leipzig, March 01 - 02 Lifestyle Many “subdivisions” LOHAS: “Lifestyles of Health and Sustainability“ LOVOS: “Lifestyles of Voluntary Simplicity“ SLOHAS: “Slow Lifestyles of Happiness and Sustainability” PARKOS: “Partizipative Konsumenten“ ……. ……. ……. AGAH Annual Meeting 2012, Leipzig, March 01 - 02 Lifestyle drugs Lifestyle drug is an imprecise term commonly applied to medications which treat non-life threatening and non-painful conditions such as baldness, impotence, wrinkles, or acne, without any medical relevance at all or only minor medical relevance relative to others. Desire for increase of personal well-being and quality of life It is sometimes intended as a pejorative, bearing the implication that the scarce medical research resources allocated to develop such drugs were spent frivolously when they could have been better spent researching cures for more serious medical conditions. -

Full Prescribing Information Warning: Suicidal Thoughts
tablets (SR) BUPROPION hydrochloride extended-release HIGHLIGHTS OF PRESCRIBING INFORMATION anxiety, and panic, as well as suicidal ideation, suicide attempt, and completed suicide. Observe hydrochloride extended-release tablets (SR) was reported. However, the symptoms persisted in some Table 3. Adverse Reactions Reported by at Least 1% of Subjects on Active Treatment and at a tablets (SR) may be necessary when coadministered with ritonavir, lopinavir, or efavirenz [see Clinical These highlights do not include all the information needed to use bupropion hydrochloride patients attempting to quit smoking with Bupropion hydrochloride extended-release tablets, USP cases; therefore, ongoing monitoring and supportive care should be provided until symptoms resolve. Greater Frequency than Placebo in the Comparator Trial Pharmacology (12.3)] but should not exceed the maximum recommended dose. extended-release tablets (SR) safely and effectively. See full prescribing information for (SR) for the occurrence of such symptoms and instruct them to discontinue Bupropion hydrochloride Carbamazepine, Phenobarbital, Phenytoin: While not systematically studied, these drugs extended-release tablets, USP (SR) and contact a healthcare provider if they experience such adverse The neuropsychiatric safety of Bupropion hydrochloride extended-release tablets (SR) was evaluated in Bupropion Bupropion may induce the metabolism of bupropion and may decrease bupropion exposure [see Clinical bupropion hydrochloride extended-release tablets (SR). Nicotine events. (5.2) a randomized, double-blind, active-and placebo-controlled study that included patients without a history Hydrochloride Hydrochloride Pharmacology (12.3)]. If bupropion is used concomitantly with a CYP inducer, it may be necessary Transdermal BUPROPION hydrochloride extended-release tablets (SR), for oral use • Seizure risk: The risk is dose-related. -

Spectrum of Digoxin-Induced Ocular Toxicity: a Case Report and Literature Review Delphine Renard1*, Eve Rubli2, Nathalie Voide3, François‑Xavier Borruat3 and Laura E
Renard et al. BMC Res Notes (2015) 8:368 DOI 10.1186/s13104-015-1367-6 CASE REPORT Open Access Spectrum of digoxin-induced ocular toxicity: a case report and literature review Delphine Renard1*, Eve Rubli2, Nathalie Voide3, François‑Xavier Borruat3 and Laura E. Rothuizen1 Abstract Background: Digoxin intoxication results in predominantly digestive, cardiac and neurological symptoms. This case is outstanding in that the intoxication occurred in a nonagenarian and induced severe, extensively documented visual symptoms as well as dysphagia and proprioceptive illusions. Moreover, it went undiagnosed for a whole month despite close medical follow-up, illustrating the difficulty in recognizing drug-induced effects in a polymorbid patient. Case presentation: Digoxin 0.25 mg qd for atrial fibrillation was prescribed to a 91-year-old woman with an esti‑ mated creatinine clearance of 18 ml/min. Over the following 2–3 weeks she developed nausea, vomiting and dyspha‑ gia, snowy and blurry vision, photopsia, dyschromatopsia, aggravated pre-existing formed visual hallucinations and proprioceptive illusions. She saw her family doctor twice and visited the eye clinic once until, 1 month after starting digoxin, she was admitted to the emergency room. Intoxication was confirmed by a serum digoxin level of 5.7 ng/ml (reference range 0.8–2 ng/ml). After stopping digoxin, general symptoms resolved in a few days, but visual complaints persisted. Examination by the ophthalmologist revealed decreased visual acuity in both eyes, 4/10 in the right eye (OD) and 5/10 in the left eye (OS), decreased color vision as demonstrated by a score of 1/13 in both eyes (OU) on Ishihara pseudoisochromatic plates, OS cataract, and dry age-related macular degeneration (ARMD). -

Carboxylesterases: Pharmacological Inhibition Regulated Expression and Transcriptional Involvement of Nuclear Receptors and Other Transcription Factors
CARBOXYLESTERASES: PHARMACOLOGICAL INHIBITION REGULATED EXPRESSION AND TRANSCRIPTIONAL INVOLVEMENT OF NUCLEAR RECEPTORS AND OTHER TRANSCRIPTION FACTORS Yuanjun Shen1, Zhanquan Shi2 and Bingfang Yan2,3 Division of Pharmaceutical Sciences James L. Winkle College of Pharmacy University of Cincinnati Cincinnati, OH 45229 USA. Address Correspondence to: Dr. Bingfang Yan Division of Pharmaceutical Sciences James L. Winkle College of Pharmacy University of Cincinnati Cincinnati, OH 45229 Phone: (513) 558-6297 Fax: (513) 558-3233 E-mail: [email protected] Running Title: Regulated expression and inhibited activity of carboxylesterases FOOTNOTES 1 Current address: Pittsburgh Heart, Lung and Blood Vascular Medicine Institute, University of Pittsburgh Department of Medicine, Pittsburgh, PA 15261, USA. 2 Division of Pharmaceutical Sciences, James L. Winkle College of Pharmacy, University of Cincinnati, Cincinnati, OH 45229, USA 3 To whom correspondence should be addressed. 4 This work was supported by National Institutes of Health Grants R01GM61988 and R01EB018748. 5 Abbreviation used: CAR, Constitutive androstane receptor; CES1, carboxylesterase-1; DEC1, Differentiated embryo chondrocyte-1; 5-FU: Fluorouracil; GR, Glucocorticoid receptor; IL-6, Interlekin- 6; LPS, Lipopolysaccharides; Nrf2, Nuclear factor (erythroid-derived 2)-like 2; p53, Tumor protein p53; PXR, Pregnane X receptor Carboxylesterases (CESs, E.C.3.1.1.1) constitute a large class of enzymes that determine the therapeutic efficacy and toxicity of ester/amide drugs. Without exceptions, all mammalian species studied express multiple forms of carboxylesterases. Two human carboxylesterases, CES1 and CES2, are major contributors to hydrolytic biotransformation. Recent studies have identified therapeutic agents that potently inhibit carboxylesterases-based catalysis. Some of them are reversible whereas others irreversible. The adrenergic antagonist carvedilol, for example, reversibly inhibits CES2 but this carboxylesterase is irreversibly inhibited by orlistat, a popular anti-obesity medicine. -

Cyproterone Acetate in the Management of Polycystic Ovary Syndrome
CORE Metadata, citation and similar papers at core.ac.uk Provided by Journal of Rawalpindi Medical College Journal of Rawalpindi Medical College (JRMC); 2018;22(3): 262-265 Original Article Comparison of Metformin and Ethinyl Estradiol- Cyproterone Acetate in the Management of Polycystic Ovary Syndrome Sadaf Ajaib, Maimoona Ali Department of Gyanecology and Obstetrics, Military Hospital, Rawalpindi Abstract predisposition. The rudimentary quandary is in the hypothalamic pituitary axis leading to incremented Background: To compare ethinyl estradiol- LH/FSH ratio1. The cumulation of anovulation and cyproterone acetate and metformin for polycystic hyperandrogenism denotes the classic form of PCOS ovarian syndrome in terms of mean hirsutism score which exhibits the adverse metabolic phenotype of the and waist hip ratio (WHR). syndrome. Clinical and biochemical features of these Methods: A total of 120 patients (60 patients in patients may vary well-according to race, ethnicity and each group) were included in the study. Group-A the diagnostic criteria used. 2 Hyper-androgenism received metformin 500mg and group-B was given may result in acne and hirsutism. Hirsutism afflicts 5- ethinyl estradiol 35 mg as treatment of polycystic 15% of females and is a paramount denouement of ovary disease (PCOD), mean hirsuitism and waist to background hyper-androgenemia. The manifestations hip ratio was measured at 6months. Hirsuitism was of polycystic ovary syndrome depends largely on measured according to Ferriman Gallway score. obesity, which exacerbates the reproductive and Circumferences were measured with an metabolic aberrations in women tormented by PCOs. anthropometric tape over light clothing. Waist girth Further studies conclude that obesity might promote was measured at the minimum circumference the phenotypic expression of PCOS in a population at between the iliac crest and the rib cage and hip girth risk of this disease.1,2 at the maximum width and their ratio was calculated Well known risk determinants for cardiovascular as baseline and after 6 months of treatment. -

(12) Patent Application Publication (10) Pub. No.: US 2012/0022039 A1 Schwink Et Al
US 20120022039A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0022039 A1 Schwink et al. (43) Pub. Date: Jan. 26, 2012 (54) NOVEL SUBSTITUTED INDANES, METHOD Publication Classification FOR THE PRODUCTION THEREOF, AND USE (51) Int. Cl. THEREOF AS DRUGS A 6LX 3L/397 (2006.01) C07D 207/06 (2006.01) C07D 2L/22 (2006.01) C07D 22.3/04 (2006.01) (75) Inventors: Lothar Schwink, Frankfurt am C07D 22L/22 (2006.01) Main (DE); Siegfried Stengelin, C07C 235/54 (2006.01) Frankfurt am Main (DE); Matthias C07D 307/14 (2006.01) Gossel, Frankfurt am Main (DE); A63L/35 (2006.01) Klaus Wirth, Frankfurt am Main A6II 3/40 (2006.01) (DE) A6II 3/445 (2006.01) A6II 3/55 (2006.01) A63L/439 (2006.01) A6II 3/66 (2006.01) (73) Assignee: SANOFI, Paris (FR) A6II 3/34 (2006.01) A6IP3/10 (2006.01) A6IP3/04 (2006.01) A6IP3/06 (2006.01) (21) Appl. No.: 13/201410 A6IPL/I6 (2006.01) A6IP3/00 (2006.01) C07D 309/04 (2006.01) (52) U.S. Cl. .................... 514/210.01; 549/426: 548/578; (22) PCT Filed: Feb. 12, 2010 546/205; 540/484; 546/112:564/176; 549/494; 514/459:514/408: 514/319; 514/212.01; 514/299; 514/622:514/471 (86). PCT No.: PCT/EP2010/051796 (57) ABSTRACT The invention relates to substituted indanes and derivatives S371 (c)(1), thereof, to physiologically acceptable salts and physiologi (2), (4) Date: Oct. 4, 2011 cally functional derivatives thereof, to the production thereof, to drugs containing at least one substituted indane according (30) Foreign Application Priority Data to the invention or derivative thereof, and to the use of the Substituted indanes according to the invention and to deriva Feb. -

Guideline for Preoperative Medication Management
Guideline: Preoperative Medication Management Guideline for Preoperative Medication Management Purpose of Guideline: To provide guidance to physicians, advanced practice providers (APPs), pharmacists, and nurses regarding medication management in the preoperative setting. Background: Appropriate perioperative medication management is essential to ensure positive surgical outcomes and prevent medication misadventures.1 Results from a prospective analysis of 1,025 patients admitted to a general surgical unit concluded that patients on at least one medication for a chronic disease are 2.7 times more likely to experience surgical complications compared with those not taking any medications. As the aging population requires more medication use and the availability of various nonprescription medications continues to increase, so does the risk of polypharmacy and the need for perioperative medication guidance.2 There are no well-designed trials to support evidence-based recommendations for perioperative medication management; however, general principles and best practice approaches are available. General considerations for perioperative medication management include a thorough medication history, understanding of the medication pharmacokinetics and potential for withdrawal symptoms, understanding the risks associated with the surgical procedure and the risks of medication discontinuation based on the intended indication. Clinical judgement must be exercised, especially if medication pharmacokinetics are not predictable or there are significant risks associated with inappropriate medication withdrawal (eg, tolerance) or continuation (eg, postsurgical infection).2 Clinical Assessment: Prior to instructing the patient on preoperative medication management, completion of a thorough medication history is recommended – including all information on prescription medications, over-the-counter medications, “as needed” medications, vitamins, supplements, and herbal medications. Allergies should also be verified and documented. -
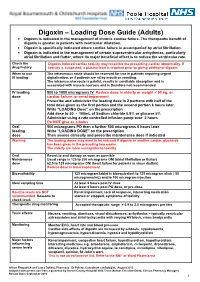
Digoxin – Loading Dose Guide (Adults) Digoxin Is Indicated in the Management of Chronic Cardiac Failure
Digoxin – Loading Dose Guide (Adults) Digoxin is indicated in the management of chronic cardiac failure. The therapeutic benefit of digoxin is greater in patients with ventricular dilatation. Digoxin is specifically indicated where cardiac failure is accompanied by atrial fibrillation. Digoxin is indicated in the management of certain supraventricular arrhythmias, particularly atrial fibrillation and flutter, where its major beneficial effect is to reduce the ventricular rate. Check the Digoxin-induced cardiac toxicity may resemble the presenting cardiac abnormality. If drug history toxicity is suspected, a plasma level is required prior to giving additional digoxin. When to use The intravenous route should be reserved for use in patients requiring urgent IV loading digitalisation, or if patients are nil by mouth or vomiting. The intramuscular route is painful, results in unreliable absorption and is associated with muscle necrosis and is therefore not recommended. IV loading 500 to 1000 micrograms IV Reduce dose in elderly or weight < 50 kg, or dose cardiac failure, or renal impairment Prescribe and administer the loading dose in 2 portions with half of the total dose given as the first portion and the second portion 6 hours later. Write “LOADING Dose” on the prescription Add dose to 50 - 100mL of Sodium chloride 0.9% or glucose 5% Administer using a rate controlled infusion pump over 2 hours Do NOT give as a bolus Oral 500 micrograms PO then a further 500 micrograms 6 hours later loading Write “LOADING DOSE” on the prescription dose Then assess clinically and prescribe maintenance dose if indicated Warning The loading doses may need to be reduced if digoxin or another cardiac glycoside has been given in the preceding two weeks. -

PRODUCT MONOGRAPH Pr WELLBUTRIN XL Bupropion
PRODUCT MONOGRAPH Pr WELLBUTRIN XL Bupropion Hydrochloride Extended-Release Tablets, USP 150 mg and 300 mg Antidepressant Name: Date of Revision: Valeant Canada LP March 20, 2017 Address: 2150 St-Elzear Blvd. West Laval, QC, H7L 4A8 Control Number: 200959 Table of Contents Page PART I: HEALTH PROFESSIONAL INFORMATION .............................................................. 2 SUMMARY PRODUCT INFORMATION ................................................................................. 2 INDICATIONS AND CLINICAL USE ....................................................................................... 2 CONTRAINDICATIONS ............................................................................................................ 3 WARNINGS AND PRECAUTIONS ........................................................................................... 3 ADVERSE REACTIONS ............................................................................................................. 9 DRUG INTERACTIONS ........................................................................................................... 19 DOSAGE AND ADMINISTRATION ....................................................................................... 22 OVERDOSAGE ......................................................................................................................... 24 ACTION AND CLINICAL PHARMACOLOGY ..................................................................... 25 STORAGE AND STABILITY .................................................................................................. -

Treatment for Calcium Channel Blocker Poisoning: a Systematic Review
Clinical Toxicology (2014), 52, 926–944 Copyright © 2014 Informa Healthcare USA, Inc. ISSN: 1556-3650 print / 1556-9519 online DOI: 10.3109/15563650.2014.965827 REVIEW ARTICLE Treatment for calcium channel blocker poisoning: A systematic review M. ST-ONGE , 1,2,3 P.-A. DUB É , 4,5,6 S. GOSSELIN ,7,8,9 C. GUIMONT , 10 J. GODWIN , 1,3 P. M. ARCHAMBAULT , 11,12,13,14 J.-M. CHAUNY , 15,16 A. J. FRENETTE , 15,17 M. DARVEAU , 18 N. LE SAGE , 10,14 J. POITRAS , 11,12 J. PROVENCHER , 19 D. N. JUURLINK , 1,20,21 and R. BLAIS 7 1 Ontario and Manitoba Poison Centre, Toronto, ON, Canada 2 Institute of Medical Science, University of Toronto, Toronto, ON, Canada 3 Department of Clinical Pharmacology and Toxicology, University of Toronto, Toronto, ON, Canada 4 Direction of Environmental Health and Toxicology, Institut national de sant é publique du Qu é bec, Qu é bec, QC, Canada 5 Centre Hospitalier Universitaire de Qu é bec, Qu é bec, QC, Canada 6 Faculty of Pharmacy, Université Laval, Qu é bec, QC, Canada 7 Centre antipoison du Qu é bec, Qu é bec, QC, Canada 8 Department of Medicine, McGill University, Montr é al, QC, Canada 9 Toxicology Consulting Service, McGill University Health Centre, Montr é al, QC, Canada 10 Centre Hospitalier Universitaire de Qu é bec, Qu é bec, QC, Canada 11 Centre de sant é et services sociaux Alphonse-Desjardins (CHAU de Lévis), L é vis, QC, Canada 12 Department of Family Medicine and Emergency Medicine, Universit é Laval, Québec, QC, Canada 13 Division de soins intensifs, Universit é Laval, Qu é bec, QC, Canada 14 Populations