Critical Assessment of Metagenome Interpretation – a Benchmark of Computational Metagenomics Software
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aspects of the Biology and Taxonomy of British Myrmecophilous Root Aphids
Aspects of the biology and taxonomy of British myrmecophilous root aphids. by Richard George PAUL, BoSc.(Lond.),17.90.(Glcsgow). Thesis submitted for the Degree of Doctor of Philosopk,. of the University of London and for the Diploma of Imperial Colleao Decemi)or 1977. Dopartaent o:vv. .;:iotory)c Cromwell Road. 1.0;AMOH )t=d41/: -1- ABSTRACT. This thesis concerns the biology and taxonomy of root feeding aphids associated with British ants. A root aphid for the purposes of this thesis is defined as an aphid which, during at least part of its life cycle feeds either (a) beneath the normal soil surface or (b) beneath a tent of soil that has been placed over it by ants. The taxonomy of the genera Paranoecia and Anoecia has been revised and some synonomies proposed. Chromosome numbers have been discovered for Anoecia spp. and are used to clarify the taxonomy. The biology of Anoecia species has been studied and new facts about their life cycles have been discovered. A key is given to the British r European, African and North American species of Anoeciinae and this is included in a key to British myrmecophilous root aphids. Suction trap catches (1968-1976) from about twenty British traps have been used as a record of seasonal flight patterns for root aphids. All the Anoecia species caught in 1975 and 1976 were identified on the basis of new taxonomic work. Catches which had formerly all been identified as Anoecia corni were found to be A. corni, A. varans and A. furcata. The information derived from the catches was used to plot relative abundance and distribution maps for the three species. -

Transatlantic Migration and the Politics of Belonging, 1919-1939
W&M ScholarWorks Dissertations, Theses, and Masters Projects Theses, Dissertations, & Master Projects Summer 2016 Between Third Reich and American Way: Transatlantic Migration and the Politics of Belonging, 1919-1939 Christian Wilbers College of William and Mary - Arts & Sciences, [email protected] Follow this and additional works at: https://scholarworks.wm.edu/etd Part of the American Studies Commons Recommended Citation Wilbers, Christian, "Between Third Reich and American Way: Transatlantic Migration and the Politics of Belonging, 1919-1939" (2016). Dissertations, Theses, and Masters Projects. Paper 1499449834. http://doi.org/10.21220/S2JD4P This Dissertation is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Dissertations, Theses, and Masters Projects by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. Between Third Reich and American Way: Transatlantic Migration and the Politics of Belonging, 1919-1939 Christian Arne Wilbers Leer, Germany M.A. University of Münster, Germany, 2006 A Dissertation presented to the Graduate Faculty of the College of William and Mary in Candidacy for the Degree of Doctor of Philosophy American Studies Program The College of William and Mary August 2016 © Copyright by Christian A. Wilbers 2016 ABSTRACT Historians consider the years between World War I and World War II to be a period of decline for German America. This dissertation complicates that argument by applying a transnational framework to the history of German immigration to the United States, particularly the period between 1919 and 1939. The author argues that contrary to previous accounts of that period, German migrants continued to be invested in the homeland through a variety of public and private relationships that changed the ways in which they thought about themselves as Germans and Americans. -

A Benchmark of Computational Metagenomics Software
bioRxiv preprint doi: https://doi.org/10.1101/099127; this version posted June 12, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Critical Assessment of Metagenome Interpretation – a benchmark of computational metagenomics software Alexander Sczyrba1*,Peter Hofmann2,3*, Peter Belmann1,3*, David Koslicki4, Stefan Janssen3,5, Johannes Dröge2,3, Ivan Gregor2,3,6, Stephan Majda2,7, Jessika Fiedler2,3, Eik Dahms2,3, Andreas Bremges1,3,8, Adrian Fritz3, Ruben Garrido-Oter2,3,9,10, Tue Sparholt Jørgensen11,12,13, Nicole Shapiro14, Philip D. Blood15, Alexey Gurevich16, Yang Bai9,17, Dmitrij Turaev18, Matthew Z. DeMaere19, Rayan Chikhi20,21, Niranjan Nagarajan22, Christopher Quince23, Fernando Meyer3, Monika Balvočiūtė24, Lars Hestbjerg Hansen11, Søren J. Sørensen12, Burton K. H. Chia22, Bertrand Denis22, Jeff L. Froula14, Zhong Wang14, Robert Egan14, Dongwan Don Kang14, Jeffrey J. Cook25, Charles Deltel26,27, Michael Beckstette28, Claire Lemaitre26,27, Pierre Peterlongo26,27, Guillaume Rizk27,29, Dominique Lavenier21,27, Yu-Wei Wu30,31, Steven W. Singer30,32, Chirag Jain33, Marc Strous34, Heiner Klingenberg35, Peter Meinicke35, Michael Barton14, Thomas Lingner36, Hsin-Hung Lin37, Yu-Chieh Liao37, Genivaldo Gueiros Z. Silva38, Daniel A. Cuevas38, Robert A. Edwards38, Surya Saha39, Vitor C. Piro40,41, Bernhard Y. Renard40, Mihai Pop42, Hans-Peter Klenk43, Markus Göker44, Nikos C. Kyrpides14, Tanja Woyke14, Julia A. Vorholt45, Paul Schulze-Lefert9,10, Edward M. Rubin14, Aaron E. Darling19, Thomas Rattei18, Alice C. McHardy2,3,10 1. Faculty of Technology and Center for Biotechnology, Bielefeld University, Bielefeld, 33594 Germany 2. -

Copyright by Louis Burrell Carrick
COPYRIGHT BY LOUIS BURRELL CARRICK 1957 A STUDY OP HYDRAS IN LAKE ERIE Contribution toward a Natural History of the Great Lakes Hydridae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of philosophy in the Graduate School of The Ohio State University By LOUIS BURRELL CARRICK, B. A., M. S. •SKHHKHS- The Ohio State University 19 £6 Approved by: Advisor / Department of Zoology and Entomology TABLE OP CONTENTS INTRODUCTION .......................................... 1 I. PROBLEMS OP TAXONOMY ................. 7 1. CRITERIA FOR THE GE N E R A ............... 7 2. METHODS USED FOR SPECIES DETERMINATION .... 11 3. IDENTIFICATION OP THE. LAKE ERIE SPECIES . 16 (a) Hydra ollgactis Pallas, 1776 ............ 17 (b) Hydra pseudollgaotis (Hyman, 1931) .... 20 (c) Hydra amerlcana Hyman, 1929 21 (d) Hydra littoralls Hyman, 1931 . .. 23 (e) Hydra came a L. Agassiz, l8jp0 .... 2I4. II. HABITATS AND DISTRIBUTION IN THE GREAT LAKES . 27 1. ABIOTIC HABITAT FACTORS AFFECTING DISTRIBUTION ................................ 29 2. DISCUSSION OF HABITATS AND DISTRIBUTION- RECORDS .............................. 32 (a) Aggregations on N e t s ..................... 34 (b) occurrence in the Plankton ............... 47 (c) Deep-Water Communities................... 49 (d) Vegetation Zones ......................... 54 (e) Lake Erie Island Ponds ................... 60 (f) Wave-swept S h o r e s ....................... 65 - ii - III. COMMUNITY INTERACTIONS WITH SPECIAL REFERENCE TO H. LITTORALIS............................. 67 1. METHODS USED IN THE STUDY A R E A ........ 69 2. SEASONAL ABUNDANCE ..................... 8l (a) The Annual C y c l e .................. 86 (b) Reproductive Potential under Culture C o n d i t i o n s ........................... 95 (c) Survival under Adverse Conditions .... 108 3. CHANGING AGGREGATIONS IN THE MICROHABITATS. -

The US National Tick Collection (USNTC)
The U.S. National Tick Collection (USNTC) For more information regarding the tick collection, contact Colleen Evans, Collections Manager, [email protected]. With over 125,000 accessioned lots, over one million specimens, their associated data, and an extensive library (reprints, monographs, and books), the U.S. National Tick collection is one of the largest curated tick collections in the World, if not the largest. It belongs to the U. S. National Museum of Natural History (Smithsonian Institution) and has been housed at Georgia Southern University since 1990. The collection contains specimens from all continents, most of the approximately 860 known species of ticks, and a quarter of the primary tick types. Ticks (Ixodida) are haematophagous mites (Acari). During their blood meal, ticks can acquire and/or transmit a number of microorganisms, some of which can cause diseases in humans, domestic and wild animals. Because of the impact of ticks on human and veterinary medicine, the USNTC is not only a significant resource of material and information for tick taxonomists but also for public health officials. History At the very beginning of the last century, while studying Rocky Mountain spotted fever outbreaks in Montana and Idaho, a team led by Dr. Howard T. Ricketts discovered the role of ticks in transmitting pathogens to humans. This finding galvanized the interest of scientists in tick systematics. The historical core of the present collection was maintained by Dr. Robert A. Cooley (1873-1968) and one of his students, Dr. Glen M. Kohls, in the Department of Entomology and Zoology of Montana State College. In 1931 the collection and its curators moved to the newly funded Rocky Mountain Laboratories (RML). -

Scientific Report 2003
Scientific Report 2003 Max Planck Institute for Plant Breeding Research Cologne Scientific Report 2003 Max Planck Institute for Plant Breeding Research Cologne 1 Copyright © 2003 MPIZ Publisher: Max Planck Institute for Plant Breeding Research (Max-Planck-Institut für Züchtungsforschung) Editor: Dr. Susanne Benner Copy Editing: Dr. Ruth Willmott, BioScript Graphics and pictures: Authors of the article Portraits: Authors or Maret Kalda Cover picture: Arp Schnittger, University of Cologne, with kind help of the REM-Laboratory, Basel, Switzerland Re-colored scanning electron micrograph showing an Arabidopsis leaf hair (trichome) undergoing cell death. Wild-type trichomes do not die, and in the depicted trichome, cell death was induced by misexpressing trichome- specifically the plant homolog of p27Kip1, the cyclin-dependent kinase inhibitor KRP1. This finding opens a new link between cell-cycle and cell-death control in plants. All rights reserved. 2 TABLE OF CONTENTS Contents Page Introduction . .6 Scientific Organisation Chart . .7 Department of Plant Developmental Biology 9 DIRECTOR: GEORGE COUPLAND Control of Flowering . .10 GEORGE COUPLAND Plant Circadian Biology . .17 SETH JON DAVIS Protein Modifiers in Plants . .20 ANDREAS BACHMAIR Genes and Tools for Molecular Breeding of New Crops . .23 GUIDO JACH Basic Biological Processes in Plant Development . .25 BERND REISS Functional Analysis of Regulators of Sugar and Stress responses in Arabidopsis . .27 CSABA KONCZ Department of Plant Microbe Interactions 31 DIRECTOR: PAUL SCHULZE-LEFERT Recognition and Signalling in Plant Disease Resistance . .32 PAUL SCHULZE-LEFERT Pathogen Defence-Related Promoter Elements and Secondary Products . .36 KLAUS HAHLBROCK Functional Analysis of Plant Defence Responses . .39 ERICH KOMBRINK Transcriptional Regulation of Defence Genes via WRKY Transcription Factors . -

Book of Abstracts DUENE E.V
The utilization of wetland biomass offers promi- sing new potential as renewable raw material and fuel. It may combine the substitution of fossil resources, sustainable land use, nature International Conference on conservation and the maintenance of ecosys- the Utilization of Emergent Wetland Plants tem services. Numerous research and development activities world wide reflect the awareness of this potential. Reed as a The international conference on the utilization of emergent wetland Renewable ResouRce plants brought together various actors from research, governance and February 14th - 16th 2013 practice in order to build networks, detect research demands and ex- Alfried Krupp Wissenschaftskolleg, change experience and information. Greifswald, Germany Abstracts of the International Conference „Reed as a Renewable 2013 Resource“ Abstracts of the International Conference Book of Abstracts DUENE e.V. D ENE at the Institute of Botany U and Landscape Ecology Alfried Krupp Wissenschaftskolleg Greifswald with the kind support of Alfried Krupp Wissenschaftskolleg Greifswald in preparation: „Paludiculture - productive management of wet peatlands“ Contribute to climate and nature to be released in September 2013 protection: MoorFutures! Ministerium für Landwirtschaft Umwelt und Verbraucherschutz offset your carbon emissions by peatland rewetting www.moorfutures.de INTERNATIONAL CONFERENCE ON THE UTILIZATION OF EMERGENT WETLAND PLANTS Reed as a Renewable Resource SCOPE The use of wetland biomass offers many opportunities to address the increasing and di- verse demand for biomass. Wetland biomass can substitute fossil resources as a raw mate- rial for industry and energy production, using both traditional and new processing lines and techniques. The cultivation and exploitation of reeds like common reed, sedges, reed canary grass, cattail, etc. -

Rhipicephalus (Boophilus) Microplus and Rhipicephalus Appendiculatus Ticks and Determination of the Expression Profile of Bm86
001-008 - Title page & table of contents 17x24 020810.pdf 1 2-8-2010 20:42:11 A contribution to the development of anti-tick vaccines A.M. Nijhof, 2010 PhD thesis Utrecht University, the Netherlands Printed by Atalanta Drukwerkbemiddeling, Houten ISBN 978-90-393-5376-9 001-008 - Title page & table of contents 17x24 020810.pdf 2 2-8-2010 20:42:11 A contribution to the development of anti-tick vaccines Een bijdrage aan de ontwikkeling van vaccins tegen teken (met een samenvatting in het Nederlands) Proefschrift ter verkrijging van de graad van doctor aan de Universiteit Utrecht op gezag van de rector magnificus, prof. dr. J.C. Stoof, ingevolge het besluit van het college voor promoties in het openbaar te verdedigen op dinsdag 7 september 2010 des middags te 12.45 uur door Ard Menzo Nijhof geboren op 24 maart 1978 te Zeist 001-008 - Title page & table of contents 17x24 020810.pdf 3 2-8-2010 20:42:11 Promotoren: Prof. dr. F. Jongejan Prof. dr. J.P.M. van Putten This research described in this thesis was financially supported by the Wellcome Trust under the 'Animal Health in the Developing World' initiative through project 075799 entitled 'Adapting recombinant anti-tick vaccines to livestock in Africa'. 001-008 - Title page & table of contents 17x24 020810.pdf 4 2-8-2010 20:42:11 In liefdevolle herinnering aan Bert Nijhof (1946-1998) 001-008 - Title page & table of contents 17x24 020810.pdf 5 2-8-2010 20:42:11 001-008 - Title page & table of contents 17x24 020810.pdf 6 2-8-2010 20:42:11 Table of contents Chapter 1 General introduction 9 Chapter -
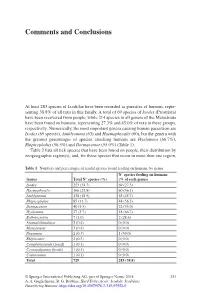
Comments and Conclusions
Comments and Conclusions At least 283 species of Ixodidae have been recorded as parasites of humans, repre- senting 38.8% of all taxa in this family. A total of 69 species of Ixodes (Prostriata) have been recovered from people, while 214 species in all genera of the Metastriata have been found on humans, representing 27.3% and 45.0% of taxa in these groups, respectively. Numerically, the most important genera causing human parasitism are Ixodes (69 species), Amblyomma (63) and Haemaphysalis (60), but the genera with the greatest percentages of species attacking humans are Hyalomma (66.7%), Rhipicephalus (56.5%) and Dermacentor (55.0%) (Table 1). Table 2 lists all tick species that have been found on people, their distribution by zoogeographic region(s), and, for those species that occur in more than one region, Table 1 Numbers and percentages of ixodid species found feeding on humans, by genus. N° species feeding on humans Genus Total N° species (%) (% of each genus) Ixodes 253 (34.7) 69 (27.3) Haemaphysalis 166 (22.8) 60 (36.1) Amblyomma 138 (18.9) 63 (45.7) Rhipicephalus 85 (11.7) 48 (56.5) Dermacentor 40 (5.5) 22 (55.0) Hyalomma 27 (3.7) 18 (66.7) Bothriocroton 7 (1.0) 2 (28.6) Anomalohimalaya 3 (0.4) 0 (0.0) Margaropus 3 (0.4) 0 (0.0) Nosomma 2 (0.3) 1 (50.0) Rhipicentor 2 (0.3) 0 (0.0) Compluriscutula (fossil) 1 (0.1) 0 (0.0) Cornupalpatum (fossil) 1 (0.1) 0 (0.0) Cosmiomma 1 (0.1) 0 (0.0) Total 729 283 (38.8) © Springer International Publishing AG, part of Springer Nature 2018 231 A. -

28Th NPS Abstract Book Cover
28th New Phytologist Symposium Functions and ecology of the plant microbiome Aldemar Hotel, Rhodes, Greece Scientific Organizing Committee Jeff Dangl (University of North Carolina, Chapel Hill, USA) Paul Schulze-Lefert (Max Planck Institute for Plant Breeding Research, Cologne, Germany) New Phytologist Organization Helen Pinfield-Wells (Deputy Managing Editor) Jill Brooke (Journal & Finance Administrator, New Phytologist) Holly Slater (Managing Editor) Acknowledgements The 28th New Phytologist Symposium is funded by the New Phytologist Trust New Phytologist Trust The New Phytologist Trust is a non-profit-making organization dedicated to the promotion of plant science, facilitating projects from symposia to open access for our Tansley reviews. Complete information is available at www.newphytologist.org Programme, abstracts and participant list compiled by Jill Brooke ‘Functions and ecology of the plant microbiome’ illustration by A.P.P.S., Lancaster, UK 1 Table of Contents Acknowledgements 1 Information for Delegates 3 Meeting Programme 4 Speaker Abstracts 7 Poster Abstracts 29 Participants 80 Maps 87 2 Information for Delegates Meeting Location The 28th New Phytologist Symposium will be held at the ALDEMAR Paradise Mare Hotel, Rhodes, Greece 18-21 May 2012. The main meeting will take place in the Rhodes & Patmos rooms. Catering As all inclusive guests at the Aldemar Paradise Mare hotel you should wear your wrist band at all times. Coffee breaks will be served in either the Kos & Karpathos rooms or the Conference center veranda. This is -

Zur Geschichte Der Universität Rostock 600 Jahre Traditio Et Innovatio Vorwort
Traditio et Innovatio Forschungsmagazin der Universität Rostock 15. Jahrgang | Heft 2 | 2010 | ISSN 1432-1513 | 4,50 Euro Zur Geschichte der Universität Rostock 600 Jahre Traditio et Innovatio Vorwort Liebe Leserin, lieber Leser, im Jahr 2019 wird die Universität Rostock ihr 600-jähriges Bestehen feiern. Grund genug, schon jetzt in einer Ausgabe unseres Forschungsmagazins eine kurze Vor- schau auf dieses Jubiläum zu geben. Wissenschaftlerinnen und Wissenschaftler aus verschiedenen Fachgebieten bieten Ihnen Einblicke in die Entwicklung unserer Alma Mater von einer spätmittelalterlichen Bildungseinrichtung hin zu einer national wie international anerkannten Universität von heute. Impressum Aus ganz unterschiedlichen Blickwinkeln beschreiben die Autorinnen und Autoren die wechselvolle wie bewegende Geschichte der Universität Rostock mit ihren Per- Herausgeber: Der Rektor der Universität sönlichkeiten und Wissenschaftsbereichen. Sie, liebe Leserin, lieber Leser, können unter anderem die beiden Rostocker Professoren Hans Spemann und Karl von Frisch Redaktionsleitung: Dr. Kristin Nölting näher kennenlernen, die für ihre Forschungen auf dem Gebiet der Zoologie mit dem Universität Rostock Nobelpreis ausgezeichnet wurden. Sie lesen etwas über den unschätzbaren Wert der Presse- und Kommunikationsstelle Ulmenstraße 69, 18057 Rostock Rostocker Matrikeln und die herausragende Sammlung medizinhistorischer Objekte. Fon +49(0)381 498-1012 In einem Beitrag über das Alltagsleben im 16. Jahrhundert an der Universität können Mail [email protected] Sie erfahren, dass ein Professor mit 400 Gulden im Jahr zu den Spitzenverdienern der Redaktion dieser Ausgabe: Universität gehörte und zu dieser Zeit den Studenten sogar die Kleidung und Frisur Prof. Dr. Kersten Krüger vorgeschrieben wurden. Und falls Sie sich schon einmal gefragt haben sollten, warum Wissenschaftlicher Beirat: man bei völliger Dunkelheit auch von „zappenduster“ spricht, in diesem Heft finden Sie Prof. -

Jahresbericht V13.Indd
CEPL AS Annual Report 2015 Imprint Published by CEPLAS – Cluster of Excellence on Plant Sciences Heinrich Heine University Düsseldorf 40204 Düsseldorf Editors Prof. Dr. Andreas P. M. Weber (responsible) Dr. Céline Hönl Dr. Juliane Schmid Photos p. 4/5 istock.com/© Aydın Mutlu p. 8/9 fotolia.com/© Vasiliy Koval p. 24 Thomas Wrobel p. 70/71/73/77/98 (b.) Steffen Köhler p. 82/83 Siegfried Werth p. 98 (t.) atelier caer All other pictures: © CEPLAS Layout atelier caer, Düsseldorf CEPL AS Annual Report 2015 Annual Report 2015 1. General presentation 1 2. Organisation 5 3. Research 9 3.1 Research Area A 10 3.1.1 Construction of a phylogenetic and genomic frame-work for the study of divergence of annual and perennial life cycles A1 Genotypic and phenotypic analysis of interspecific hybrids between A. montbretiana and A. alpina 11 A2 Analysis of the roles of PERPETUAL FLOWERING 1 direct target genes in perennial flowering and their divergence in sister annual species 12 A3 Comparison of annual-perennial species pairs across the Brassicaceae 12 A4 Nutrient recycling in the perennial plant Arabis alpina 13 A5 Molecular characterisation of senescence in annual and perennial plants 13 A6 Photoperiodic control of flowering time in Arabis alpina 14 3.1.2 Elucidation of regulatory networks that determine formation and identity of meristems A7 The control of adventitious root formation in the perennial Arabis alpina 15 A8 Developmental basis for asexual reproduction in Cardamine 15 A9 Mechanisms involved in perennial life cycle of Arabis alpina: The role of auxin in perennial traits 16 A10 The regulation of inflorescence branching in Arabis alpina 17 A11 The regulation of inflorescence branching in Arabis alpina 17 A12 Analysis of axillary meristem initiation in perennial plant Arabis alpina 18 A13 Genotypic and phenotypic analysis of tiller development in cultivated (Hordeum vulgare) and wild barley species (H.