Apoptotic Effects of Drug Targeting Conjugates Containing Different Gnrh Analogs on Colon Carcinoma Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Tanibirumab (CUI C3490677) Add to Cart
5/17/2018 NCI Metathesaurus Contains Exact Match Begins With Name Code Property Relationship Source ALL Advanced Search NCIm Version: 201706 Version 2.8 (using LexEVS 6.5) Home | NCIt Hierarchy | Sources | Help Suggest changes to this concept Tanibirumab (CUI C3490677) Add to Cart Table of Contents Terms & Properties Synonym Details Relationships By Source Terms & Properties Concept Unique Identifier (CUI): C3490677 NCI Thesaurus Code: C102877 (see NCI Thesaurus info) Semantic Type: Immunologic Factor Semantic Type: Amino Acid, Peptide, or Protein Semantic Type: Pharmacologic Substance NCIt Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor tyrosine kinase expressed by endothelial cells, while VEGF is overexpressed in many tumors and is correlated to tumor progression. PDQ Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor -

Synthesis and in Vitro Biochemical Evaluation of Oxime Bond-Linked
Synthesis and in vitro biochemical evaluation of oxime bond- linked daunorubicin–GnRH-III conjugates developed for targeted drug delivery Sabine Schuster1,2, Beáta Biri-Kovács1,2, Bálint Szeder3, Viktor Farkas4, László Buday3, Zsuzsanna Szabó5, Gábor Halmos5 and Gábor Mező*1,2 Full Research Paper Open Access Address: Beilstein J. Org. Chem. 2018, 14, 756–771. 1MTA-ELTE Research Group of Peptide Chemistry, Hungarian doi:10.3762/bjoc.14.64 Academy of Sciences, Eötvös L. University, 1117 Budapest, Hungary, 2Institute of Chemistry, Eötvös L. University, 1117 Budapest, Received: 12 January 2018 Hungary, 3Research Centre for Natural Sciences, Institute of Accepted: 15 March 2018 Enzymology, Hungarian Academy of Sciences, 1117 Budapest, Published: 04 April 2018 Hungary, 4MTA-ELTE Protein Modelling Research Group, Hungarian Academy of Sciences, Eötvös L. University, 1117 Budapest, Hungary This article is part of the Thematic Series "Peptide–drug conjugates". and 5Department of Biopharmacy, Faculty of Pharmacy, University of Debrecen, 4032 Debrecen, Hungary Guest Editor: N. Sewald Email: © 2018 Schuster et al.; licensee Beilstein-Institut. Gábor Mező* - [email protected] License and terms: see end of document. * Corresponding author Keywords: cytostatic effect; daunorubicin; drug-targeting; GnRH derivatives; oxime linkage Abstract Gonadotropin releasing hormone-III (GnRH-III), a native isoform of the human GnRH isolated from sea lamprey, specifically binds to GnRH receptors on cancer cells enabling its application as targeting moieties for anticancer drugs. Recently, we reported on the identification of a novel daunorubicin–GnRH-III conjugate (GnRH-III–[4Lys(Bu), 8Lys(Dau=Aoa)] with efficient in vitro and in vivo antitumor activity. To get a deeper insight into the mechanism of action of our lead compound, the cellular uptake was followed by confocal laser scanning microscopy. -

AHRQ Healthcare Horizon Scanning System – Status Update Horizon
AHRQ Healthcare Horizon Scanning System – Status Update Horizon Scanning Status Update: April 2015 Prepared for: Agency for Healthcare Research and Quality U.S. Department of Health and Human Services 540 Gaither Road Rockville, MD 20850 www.ahrq.gov Contract No. HHSA290-2010-00006-C Prepared by: ECRI Institute 5200 Butler Pike Plymouth Meeting, PA 19462 April 2015 Statement of Funding and Purpose This report incorporates data collected during implementation of the Agency for Healthcare Research and Quality (AHRQ) Healthcare Horizon Scanning System by ECRI Institute under contract to AHRQ, Rockville, MD (Contract No. HHSA290-2010-00006-C). The findings and conclusions in this document are those of the authors, who are responsible for its content, and do not necessarily represent the views of AHRQ. No statement in this report should be construed as an official position of AHRQ or of the U.S. Department of Health and Human Services. A novel intervention may not appear in this report simply because the System has not yet detected it. The list of novel interventions in the Horizon Scanning Status Update Report will change over time as new information is collected. This should not be construed as either endorsements or rejections of specific interventions. As topics are entered into the System, individual target technology reports are developed for those that appear to be closer to diffusion into practice in the United States. A representative from AHRQ served as a Contracting Officer’s Technical Representative and provided input during the implementation of the horizon scanning system. AHRQ did not directly participate in the horizon scanning, assessing the leads or topics, or provide opinions regarding potential impact of interventions. -
![1 SUPPLEMENTARY DATA Draft Medline Search – Pubmed Interface 1# "Puberty"[Mesh] OR (Puberties) OR (Pubertal Maturati](https://docslib.b-cdn.net/cover/5679/1-supplementary-data-draft-medline-search-pubmed-interface-1-puberty-mesh-or-puberties-or-pubertal-maturati-1835679.webp)
1 SUPPLEMENTARY DATA Draft Medline Search – Pubmed Interface 1# "Puberty"[Mesh] OR (Puberties) OR (Pubertal Maturati
SUPPLEMENTARY DATA Draft Medline search – PubMed interface 1# "Puberty"[Mesh] OR (Puberties) OR (Pubertal Maturation) OR (Early Puberty) OR "Puberty, Precocious"[Mesh] OR (Precocious Puberty) OR "Precocious Puberty, Central" [Supplementary Concept] OR (Central Precocious Puberty) OR "Sexual precocity" [Supplementary Concept] OR (Idiopathic sexual precocity) OR (Familial precocious puberty) OR (Sexual Precocity) OR "Sexual Maturation"[Mesh] OR (Maturation, Sexual) OR (Maturation, Sex) OR (Sex Maturation) OR "Sexual Development"[Mesh] OR (Development, Sexual) OR (Sex Development) OR (Development, Sex) OR (Gonadal Disorder Maturation) OR (Pubertal Onset) OR "Menstruation Disturbances"[Mesh] OR (Precocious Menses) OR (Early Menses) OR (Early Menarche) OR (Precocious Menarche) OR (Disorders of Puberty) OR (Gonadotropin-Dependent Precocious Puberty) OR (Gonadotropin-Independent Precocious Puberty) OR (Isolated Precocious Thelarche) OR (Isolated Precocious Pubarche) OR (Isolated Precocious Menarche) 2# "Gonadotropin-Releasing Hormone"[Mesh] OR (Gonadotropin Releasing Hormone) OR (Luteinizing Hormone-Releasing Hormone) OR (Luteinizing Hormone Releasing Hormone) OR (GnRH) OR (Gonadoliberin) OR (Gonadorelin) OR (LFRH) OR (LH-FSH Releasing Hormone) OR (LH FSH Releasing Hormone) OR (LH-Releasing Hormone) OR (LH Releasing Hormone) OR (LH-RH) OR (LHFSH Releasing Hormone) OR (Releasing Hormone, LHFSH) OR (LHFSHRH) OR (LHRH) OR (Luliberin) OR (FSH-Releasing Hormone) OR (FSH Releasing Hormone) OR (Gn-RH) OR (Factrel) OR (Gonadorelin Acetate) OR (Kryptocur) -
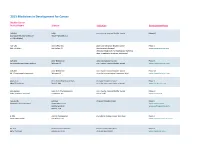
Adis R&D Insight
2015 Medicines in Development for Cancer Bladder Cancer Product Name Sponsor Indication Development Phase ABI-009 AADi non-muscle invasive bladder cancer Phase I/II (nanoparticle albumin-bound Pacific Palisades, CA mTOR inhibitor) ACP-196 Acerta Pharma platinum-refractory bladder cancer Phase II (Btk inhibitor) San Carlos, CA (combination therapy) www.acerta-pharma.com (see also head/neck, hematological, leukemia, lung, lymphoma, myeloma, pancreatic) ALT-801 Altor BioScience advanced bladder cancer, Phase II (immunotherapy fusion protein) Miramar, FL non-muscle invasive bladder cancer www.altorbioscience.com ALT-803 Altor BioScience non-muscle invasive bladder cancer Phase I/II (IL-15 superagonist complex) Miramar, FL (see also hematological, myeloma, skin) www.altorbioscience.com apatorsen OncoGenex Pharmaceuticals metastatic bladder cancer Phase II (Hsp27 inhibitor) Bothell, WA (see also lung, pancreatic, prostate) www.oncogenex.com apaziquone Spectrum Pharmaceuticals non-muscle invasive bladder cancer Phase III (DNA synthesis inhibitor) Henderson, NV (Fast Track) www.sppirx.com ASG-15ME Agensys relapsed bladder cancer Phase I (antibody drug conjugate) Santa Monica, CA www.agensys.com Seattle Genetics www.seattlegenetics.com Bothell, WA B-701 BioClin Therapeutics metastatic bladder cancer (2nd-line) Phase II (anti-FGFR3 mAb) San Ramon, CA www.bioclintherapeutics.com BC-819 BioCancell Therapeutics bladder cancer (2nd-line) Phase II (gene therapy) Jerusalem, Israel (see also pancreatic) www.biocancell.com Bladder Cancer Product Name Sponsor -

Stembook 2018.Pdf
The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 WHO/EMP/RHT/TSN/2018.1 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances. Geneva: World Health Organization; 2018 (WHO/EMP/RHT/TSN/2018.1). Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. -

(INN) for Biological and Biotechnological Substances
WHO/EMP/RHT/TSN/2019.1 International Nonproprietary Names (INN) for biological and biotechnological substances (a review) 2019 WHO/EMP/RHT/TSN/2019.1 International Nonproprietary Names (INN) for biological and biotechnological substances (a review) 2019 International Nonproprietary Names (INN) Programme Technologies Standards and Norms (TSN) Regulation of Medicines and other Health Technologies (RHT) Essential Medicines and Health Products (EMP) International Nonproprietary Names (INN) for biological and biotechnological substances (a review) FORMER DOCUMENT NUMBER: INN Working Document 05.179 © World Health Organization 2019 All rights reserved. Publications of the World Health Organization are available on the WHO website (www.who.int) or can be purchased from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications –whether for sale or for non-commercial distribution– should be addressed to WHO Press through the WHO website (www.who.int/about/licensing/copyright_form/en/index.html). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted and dashed lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. -

Market Access Right Product, Right Patient, Tight Price, Tighter Reimbursement
invivo.pharmamedtechbi.com NOVEMBER 2016 Invol. 34 ❚ no. 10 Vivopharma intelligence ❚ informa Market Access Right product, right patient, tight price, tighter reimbursement Value Frameworks Price Transparency Real-World Evidence Outcomes-Based Reimbursement invivo.pharmamedtechbi.com CONTENTS ❚ In Vivo Pharma intelligence | November 2016 26 Curative Regenerative Medicines: Preparing Health Care Systems For The Coming Wave FARAZ ALI, TED SLOCOMB AND MICHAEL WERNER We may be at the dawn of a new era of curative regenerative therapies, but their very nature may create barriers to adoption. The Alliance for Regenerative Medicine frames the opportunities and challenges for the industry, arguing that policy makers must begin to understand THE MARKET ACCESS ISSUE the ways that these therapies represent value for money. 10 22 States Fight For Drug Price US Outcomes-Based 1 Transparency Contracts: Big Uptick In ED SILVERMAN Interest, But Not Execution California’s Proposition 61 and a number CATHY KELLY of other state-led measures to control The promise of outcomes-based drug prices failed to gain traction in contracts for biopharmaceuticals has 2016. But states are likely to remain a yet to be realized in the US. Although battleground in the war against costs in precise numbers are hard to come by, a pharma-friendly Trump administration. payers and manufacturers agree the field is still nascent. However, they 16 don’t seem to be giving up on the idea. Is The UK Still Open 34 For Medtech Innovation Reimbursement? Real-World Evidence ASHLEY YEO And The Quest For Confusing and ineffective channels of European Market Access innovation adoption and a lack of FRANcescA BRUce funding hamper the UK’s reputation as Real-world evidence promises to solve one of the world’s health care capitals many problems inherent in getting a and a driver of care excellence – in spite drug to patients at a good price. -

Gnrh-R Targeted Lytic Peptide Sensitizes BRCA Wild-Type Ovarian Cancer to PARP Inhibition
Author Manuscript Published OnlineFirst on March 29, 2019; DOI: 10.1158/1535-7163.MCT-18-0770 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. GnRH-R targeted lytic peptide sensitizes BRCA wild-type ovarian cancer to PARP inhibition Shaolin Ma1,2, Sunila Pradeep1,3, Alejandro Villar-Prados1,4, Yunfei, Wen1, Emine Bayraktar1, Lingegowda S. Mangala1, Mark Seungwook Kim1, Sherry Y. Wu1, Wei Hu1, Cristian Rodriguez- Aguayo5,6, Carola Leuschner7, Xiaoyan Liang2, Prahlad T. Ram8, Katharina Schlacher5, Robert L. Coleman1 and Anil K. Sood1,5,9 1Department of Gynecologic Oncology and Reproductive Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Reproductive Medicine Research Center, the Sixth Affiliated Hospital of Sun Yat-Sen University, Guangzhou, China; 3Department of Obstetrics and Gynecology, Medical College of Wisconsin, Milwaukee, WI, 53226, USA; 4University of Puerto Rico, School of Medicine, Medical Sciences Campus, San Juan, PR, 00936, USA ; 5Department of Cancer Biology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 6Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 7VP of Research, Esperance Pharmaceuticals, 909 Fannin, Suite 2000, Houston, TX 77010-1028; 8Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 9Center for RNA Interference and Non-Coding RNA, The University of Texas MD Anderson Cancer Center, Houston, TX, USA. Correspondence: Dr. A K Sood, Department of Gynecologic Oncology and Reproductive Medicine, Unit 1362, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX 77030, USA. -

The Role of Gonadotropin-Releasing Hormone (Gnrh) in Endometrial Cancer
cells Review The Role of Gonadotropin-Releasing Hormone (GnRH) in Endometrial Cancer Günter Emons * and Carsten Gründker Department of Gynecology and Obstetrics, University Medicine Göttingen, 37075 Göttingen, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-551-396-5632 Abstract: Endometrial cancer (EC) is one of the most common gynecological malignancies. Go- nadotropin releasing hormone (GnRH) is a decapeptide first described to be secreted by the hy- pothalamus to regulate pituitary gonadotropin secretion. In this systematic review, we analyze and summarize the data indicating that most EC express GnRH and its receptor (GnRH-R) as part of an autocrine system regulating proliferation, the cell cycle, and apoptosis. We analyze the available data on the expression and function of GnRH-II, its putative receptor, and its signal transduction. GnRH-I and GnRH-II agonists, and antagonists as well as cytotoxic GnRH-I analogs, have been shown to inhibit proliferation and to induce apoptosis in human EC cell lines in pre-clinical models. Treatment with conventional doses of GnRH-agonists that suppress pituitary gonadotropin secretion and ovar- ian estrogen production has become part of fertility preserving therapy of early EC or its pre-cancer (atypical endometrial hyperplasia). Conventional doses of GnRH-agonists had marginal activity in advanced or recurrent EC. Higher doses or more potent analogs including GnRH-II antagonists have not yet been used clinically. The cytotoxic GnRH-analog Zoptarelin Doxorubicin has shown encouraging activity in a phase II trial in patients with advanced or recurrent EC, which expressed GnRH-R. In a phase III trial in patients with EC of unknown GnRH-R expression, the cytotoxic GnRH doxorubicin conjugate was not superior to free doxorubicin. -

Role of Gonadotropin-Releasing Hormone (Gnrh) in Ovarian Cancer
cells Review Role of Gonadotropin-Releasing Hormone (GnRH) in Ovarian Cancer Carsten Gründker * and Günter Emons Department of Gynecology and Obstetrics, University Medicine Göttingen, 37075 Göttingen, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-551-39-69810 Abstract: The hypothalamus–pituitary–gonadal (HPG) axis is the endocrine regulation system that controls the woman’s cycle. The gonadotropin-releasing hormone (GnRH) plays the central role. In addition to the gonadotrophic cells of the pituitary, GnRH receptors are expressed in other reproductive organs, such as the ovary and in tumors originating from the ovary. In ovarian cancer, GnRH is involved in the regulation of proliferation and metastasis. The effects on ovarian tumors can be indirect or direct. GnRH acts indirectly via the HPG axis and directly via GnRH receptors on the surface of ovarian cancer cells. In this systematic review, we will give an overview of the role of GnRH in ovarian cancer development, progression and therapy. Keywords: ovarian cancer; gonadotropin-releasing hormone; GnRH; LHRH 1. Introduction Citation: Gründker, C.; Emons, G. Ovarian carcinomas are the eighth most common malignant tumor disease in women Role of Gonadotropin-Releasing diagnosed in the world [1]. Although the risk of developing ovarian cancer in course Hormone (GnRH) in Ovarian Cancer. of their life is only 1.3% for every woman [2], ovarian cancer is the fifth most common Cells 2021, 10, 437. https://doi.org/ cancer-associated cause of death in women [3]. With a 5-year survival rate of 43%, ovarian 10.3390/cells10020437 cancer is the deadliest gynecological disease. -

Product List
PRODUCT LIST Active Pharma Ingredients API Acid Reflux Disorders Product Pharmacopoeia CAS No. Technical Document A Acotiamide Hydrochloride - 773092-05-0 - Almagate EP 66827-12-1 - Aluminum Hydroxide BP, USP 21645-51-2 - C Cimetidine - 51481-61-9 - D Dexlansoprazole - 138530-94-6 - Dexlansoprazole - 313640-86-7 DMF Sesquihydrate Dexrabeprazole Sodium - 171440-18-9 ODMF, Tech Pack E Ecabet Sodium - 86408-72-2 DMF Esomeprazole - 119141-88-7 - Esomeprazole Magneisum - 217087-10-0 - Dihydrate Esomeprazole Magnesium - 161973-10-0 - Esomeprazole Magnesium USP 217087-09-7 CEP, DMF Trihydrate Esomeprazole Potassium IHS 161796-84-5 DMF Esomeprazole Sodium - 161796-78-7 - Esomeprazole Zinc/ 793668-08-3, - - Base/Strontium 119141-88-7 F Famotidine - 76824-35-6 - E Glycopyrrolate - 596-51-0 - H Hydrotelcite BP 12304-65-3 - I Ilaprazole IHS, IP 172152-36-2 DMF Ilaprazole Sodium - 172152-50-0 ODMF, Tech Pack Itopride - 122898-67-3 - Itopride Hydrochloride IHS 122892-31-3 - Patent Disclaimer: Products protected by valid patents are not offered for sale in countries, where the sale of such products constitutes a patent infringement. The buyers should make their independent evaluation of the patent scenario for their respective markets and will be responsible for all patent related liabilities. 2021-01-11 © ExSyn Corp | All rights reserved. | www.exsyncorp.com 1 API Acid Reflux Disorders Product Pharmacopoeia CAS No. Technical Document L Lafutidine - 118288-08-7 - Lansoprazole - 103577-45-3 - Lansoprazole Sodium - 226904-00-3 - M Magaldrate USP 74978-16-8