An Investigation of Transition Metal Catalysts For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Assessment of Portable HAZMAT Sensors for First Responders
The author(s) shown below used Federal funds provided by the U.S. Department of Justice and prepared the following final report: Document Title: Assessment of Portable HAZMAT Sensors for First Responders Author(s): Chad Huffman, Ph.D., Lars Ericson, Ph.D. Document No.: 246708 Date Received: May 2014 Award Number: 2010-IJ-CX-K024 This report has not been published by the U.S. Department of Justice. To provide better customer service, NCJRS has made this Federally- funded grant report available electronically. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Assessment of Portable HAZMAT Sensors for First Responders DOJ Office of Justice Programs National Institute of Justice Sensor, Surveillance, and Biometric Technologies (SSBT) Center of Excellence (CoE) March 1, 2012 Submitted by ManTech Advanced Systems International 1000 Technology Drive, Suite 3310 Fairmont, West Virginia 26554 Telephone: (304) 368-4120 Fax: (304) 366-8096 Dr. Chad Huffman, Senior Scientist Dr. Lars Ericson, Director UNCLASSIFIED This project was supported by Award No. 2010-IJ-CX-K024, awarded by the National Institute of Justice, Office of Justice Programs, U.S. Department of Justice. The opinions, findings, and conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect those of the Department of Justice. This document is a research report submitted to the U.S. Department of Justice. This report has not been published by the Department. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. -
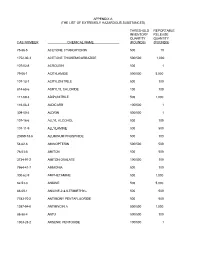
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

Cyanohydrin - Wikipedia, the Free Encyclopedia
Cyanohydrin - Wikipedia, the free encyclopedia http://en.wikipedia.org/wiki/Cyanohydrin Cyanohydrin From Wikipedia, the free encyclopedia A cyanohydrin is a functional group found in organic compounds. Cyanohydrins have the formula R2C(OH)CN, where R is H, alkyl, or aryl. Cyanohydrins are industrially important precursors to carboxylic acids and some amino acids. Cyanohydrins can be formed by the cyanohydrin reaction, which involves treating a ketone or an aldehyde with hydrogen cyanide (HCN) in the presence of excess amounts of sodium cyanide (NaCN) as a catalyst: The structure of a general RR’C=O + HCN → RR’C(OH)CN cyanohydrin. In this reaction, the nucleophilic CN− ion attacks the electrophilic carbonyl carbon in the ketone, followed by protonation by HCN, thereby regenerating the cyanide anion. Cyanohydrins are also prepared by displacement of sulfite by cyanide salts:[1] Cyanohydrins are intermediates in the Strecker amino acid synthesis. Contents 1 Acetone cyanohydrins 2 Other cyanohydrins 3 References 4 External links Acetone cyanohydrins Acetone cyanohydrin, (CH3)2C(OH)CN is the cyanohydrin of acetone. It is generated as an intermediate in the industrial production of methyl methacrylate.[2] In the laboratory, this liquid serves as a source of HCN, which is inconveniently volatile.[3] Thus, acetone cyanohydrin can be used for the preparation other cyanohydrins, for of HCN to Michael acceptors, and for the formylation of arenes. Treatment of this cyanohydrin with lithium hydride affords anhydrous lithium cyanide: 1 of 3 6/3/10 6:07 PM Cyanohydrin - Wikipedia, the free encyclopedia http://en.wikipedia.org/wiki/Cyanohydrin Other cyanohydrins Mandelonitrile, with the formula C6H5CH(OH)CN, occurs in small amounts in the pits of some fruits.[1] Related cyanogenic glycosides are known, such as amygdalin. -

Process for Producing Methyl Methacrylate Verfahren Zur Herstellung Von Methylmethacrylat Procede Pour La Fabrication De Methacrylate De Methyle
~™ II 1 1 III II II 1 1 II II I Ml II I II I II (19) J European Patent Office Office europeen des brevets (11) EP 0 406 676 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publicationation and mention (51 ) |nt. CI.6: C07C 69/54, C07C 67/20 of the grant of the patent: 27.03.1996 Bulletin 1996/13 (21) Application number: 90112194.7 (22) Date of filing: 27.06.1990 (54) Process for producing methyl methacrylate Verfahren zur Herstellung von Methylmethacrylat Procede pour la fabrication de methacrylate de methyle (84) Designated Contracting States: • Ebata, Shuji, DE ES FR GB IT NL C/o Mitsubishi Gas Chem. Com. Tayuhama, Niigata-shi, Niigata-ken (JP) (30) Priority: 04.07.1989 JP 171190/89 (74) Representative: Turk, Gille, Hrabal, Leifert (43) Date of publication of application: Brucknerstrasse 20 09.01.1991 Bulletin 1991/02 D-40593 Dusseldorf (DE) (73) Proprietor: MITSUBISHI GAS CHEMICAL (56) References cited: COMPANY, INC. DE-A- 3 436 608 Chiyoda-ku, Tokyo (JP) • PATENT ABSTRACTS OF JAPAN vol. 14, no. 68 (72) Inventors: (C- 686)(401 1 ), 8 February 1 990; & JP-A-1 290653 • Higuchi, Hirofumi, (MITSUBISHI GAS CHEM) 22.11. 1989 C/o Mitsubishi Gas Chem. Com. Tayuhama, Niigata-shi, Niigata-ken (JP) Remarks: • Kida, Koichi, The file contains technical information submitted C/o Mitsubishi Gas Chem. Com. after the application was filed and not included in this Tayuhama, Niigata-shi, Niigata-ken (JP) specification CO CO CO o Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. -

Provisional Peer-Reviewed Toxicity Values for Acetone Cyanohydrin (Casrn 75-86-5)
EPA/690/R-12/001F l Final 10-01-2012 Provisional Peer-Reviewed Toxicity Values for Acetone Cyanohydrin (CASRN 75-86-5) Superfund Health Risk Technical Support Center National Center for Environmental Assessment Office of Research and Development U.S. Environmental Protection Agency Cincinnati, OH 45268 AUTHORS, CONTRIBUTORS, AND REVIEWERS CHEMICAL MANAGER Scott C. Wesselkamper, PhD National Center for Environmental Assessment, Cincinnati, OH DRAFT DOCUMENT PREPARED BY ICF International 9300 Lee Highway Fairfax, VA 22031 PRIMARY INTERNAL REVIEWERS Zheng (Jenny) Li, PhD, DABT National Center for Environmental Assessment, Washington, DC Anuradha Mudipalli, MSc, PhD National Center for Environmental Assessment, Research Triangle Park, NC This document was externally peer reviewed under contract to Eastern Research Group, Inc. 110 Hartwell Avenue Lexington, MA 02421-3136 Questions regarding the contents of this document may be directed to the U.S. EPA Office of Research and Development’s National Center for Environmental Assessment, Superfund Health Risk Technical Support Center (513-569-7300). ii Acetone cyanohydrin TABLE OF CONTENTS COMMONLY USED ABBREVIATIONS ................................................................................... iv BACKGROUND .............................................................................................................................1 DISCLAIMERS ...............................................................................................................................1 QUESTIONS REGARDING -
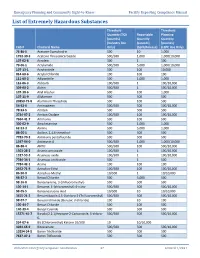
List of Extremely Hazardous Substances
Emergency Planning and Community Right-to-Know Facility Reporting Compliance Manual List of Extremely Hazardous Substances Threshold Threshold Quantity (TQ) Reportable Planning (pounds) Quantity Quantity (Industry Use (pounds) (pounds) CAS # Chemical Name Only) (Spill/Release) (LEPC Use Only) 75-86-5 Acetone Cyanohydrin 500 10 1,000 1752-30-3 Acetone Thiosemicarbazide 500/500 1,000 1,000/10,000 107-02-8 Acrolein 500 1 500 79-06-1 Acrylamide 500/500 5,000 1,000/10,000 107-13-1 Acrylonitrile 500 100 10,000 814-68-6 Acrylyl Chloride 100 100 100 111-69-3 Adiponitrile 500 1,000 1,000 116-06-3 Aldicarb 100/500 1 100/10,000 309-00-2 Aldrin 500/500 1 500/10,000 107-18-6 Allyl Alcohol 500 100 1,000 107-11-9 Allylamine 500 500 500 20859-73-8 Aluminum Phosphide 500 100 500 54-62-6 Aminopterin 500/500 500 500/10,000 78-53-5 Amiton 500 500 500 3734-97-2 Amiton Oxalate 100/500 100 100/10,000 7664-41-7 Ammonia 500 100 500 300-62-9 Amphetamine 500 1,000 1,000 62-53-3 Aniline 500 5,000 1,000 88-05-1 Aniline, 2,4,6-trimethyl- 500 500 500 7783-70-2 Antimony pentafluoride 500 500 500 1397-94-0 Antimycin A 500/500 1,000 1,000/10,000 86-88-4 ANTU 500/500 100 500/10,000 1303-28-2 Arsenic pentoxide 100/500 1 100/10,000 1327-53-3 Arsenous oxide 100/500 1 100/10,000 7784-34-1 Arsenous trichloride 500 1 500 7784-42-1 Arsine 100 100 100 2642-71-9 Azinphos-Ethyl 100/500 100 100/10,000 86-50-0 Azinphos-Methyl 10/500 1 10/10,000 98-87-3 Benzal Chloride 500 5,000 500 98-16-8 Benzenamine, 3-(trifluoromethyl)- 500 500 500 100-14-1 Benzene, 1-(chloromethyl)-4-nitro- 500/500 -

Hydrogen Cyanide and Cyanides: Human Health Aspects
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization, or the World Health Organization. Concise International Chemical Assessment Document 61 HYDROGEN CYANIDE AND CYANIDES: HUMAN HEALTH ASPECTS Please note that the layout and pagination of this pdf file are not identical to the version in press First draft prepared by Prof. Fina Petrova Simeonova, Consultant, National Center of Hygiene, Medical Ecology and Nutrition, Sofia, Bulgaria; and Dr Lawrence Fishbein, Fairfax, Virginia, USA Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 2004 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management -

1 Draft Chemicals (Management and Safety)
Draft Chemicals (Management and Safety) Rules, 20xx In exercise of the powers conferred by Sections 3, 6 and 25 of the Environment (Protection) Act, 1986 (29 of 1986), and in supersession of the Manufacture, Storage and Import of Hazardous Chemical Rules, 1989 and the Chemical Accidents (Emergency Planning. Preparedness and Response) Rules, 1996, except things done or omitted to be done before such supersession, the Central Government hereby makes the following Rules relating to the management and safety of chemicals, namely: 1. Short Title and Commencement (1) These Rules may be called the Chemicals (Management and Safety) Rules, 20xx. (2) These Rules shall come into force on the date of their publication in the Official Gazette. Chapter I Definitions, Objectives and Scope 2. Definitions (1) In these Rules, unless the context otherwise requires (a) “Act” means the Environment (Protection) Act, 1986 (29 of 1986) as amended from time to time; (b) “Article” means any object whose function is determined by its shape, surface or design to a greater degree than its chemical composition; (c) “Authorised Representative” means a natural or juristic person in India who is authorised by a foreign Manufacturer under Rule 6(2); (d) “Chemical Accident” means an accident involving a sudden or unintended occurrence while handling any Hazardous Chemical, resulting in exposure (continuous, intermittent or repeated) to the Hazardous Chemical causing death or injury to any person or damage to any property, but does not include an accident by reason only -

Material Safety Data Sheet ______
1 _____________________________________________________________________________ Material Safety Data Sheet _____________________________________________________________________________ MSDS1004 revised 20-AUG-2007 ACETONE CYANOHYDRIN _____________________________________________________________________________ CHEMICAL PRODUCT/COMPANY IDENTIFICATION _____________________________________________________________________________ Material Identification MSDS Number : 1004 CAS Number : 75-86-5 Formula : CH3C(CH3)OH)CN Molecular Weight : 85.11 CAS Name : PROPANENITRILE, 2-HYDROXY-2-METHYL Trade names and Synonyms 2-HYDROXY-2-METHYLPROPANENITRILE 2-METHYLLACTONITRILE a-HYDROXYISOBUTYRONITRILE 2-HYDROXYISOBUTYRONITRILE Company Identification MANUFACTURER/DISTRIBUTOR Lucite International, Inc. 7275 Goodlett Farms Parkway Cordova, TN 38018 PHONE NUMBERS Product Information : 1-800-4-LUCITE Transportation Emergency : 1-800-424-9300 Medical Emergency : 1-877-886-2143 _____________________________________________________________________________ COMPOSITION/INFORMATION ON INGREDIENTS _____________________________________________________________________________ Components Material CAS Number % ACETONE CYANOHYDRIN 75-86-5 97.5 HYDROGEN CYANIDE 74-90-8 0.1 ACETONE 67-64-1 0.9 STABILIZER AND INERTS* 1.1 WATER 7732-18-5 0.4 Components (Remarks) *Sulfate salts of trimethylamine 2 _____________________________________________________________________________ HAZARDS IDENTIFICATION _____________________________________________________________________________ -

Acetone Cyanohydrin 2506
ACETONE CYANOHYDRIN 2506 (CH3)2C(OH)CN MW: 85.11 CAS: 75-86-5 RTECS: OD9275000 METHOD: 2506, Issue 2 EVALUATION: PARTIAL Issue 1: 15 May 1985 Issue 2: 15 August 1994 OSHA: no PEL PROPERTIES: liquid; d 0.932 g/mL @ 19 °C; BP 95 °C; MP −19 NIOSH: C 1 ppm/15 min °C; VP 110 Pa (0.8 mm Hg) @ 20 °C ACGIH: no TLV (1 ppm = 3.48 mg/m³ SYNONYMS: 2-cyano-2-propanol; 2-methyllactonitrile; 2-hydroxy-2-methylpropanenitrile SAMPLING MEASUREMENT SAMPLER: SOLID SORBENT TUBE TECHNIQUE: GAS CHROMATOGRAPH, NP (Porapak QS, 100 mg/50 mg) (THERMIONIC) DETECTOR FLOW RATE: 0.2 L/min ANALYTE: acetone cyanohydrin VOL-MIN: 0.3 L @ 1 ppm DESORPTION: 1 mL ethyl acetate; ultrasonicate 60 min -MAX: 12 L INJECTION SHIPMENT: in refrigerated container @ 0 °C VOLUME: 5 µL, 1 µL flush SAMPLE TEMPERATURE-INJECTION: 100 °C STABILITY: at least 5 days if stored at @ 0 °C immediately -DETECTOR: 100 °C following collection -COLUMN: 70 °C BLANKS: 2 to 10 field blanks per set CARRIER GAS: N2, 33 mL/min ACCURACY COLUMN: PTFE, 2 m × 3.2-mm OD, packed with 5% OV-17 on 40/60 mesh Chromosorb T RANGE STUDIED: 0.33 to 16.7 mg/m³ [1] (APPENDIX) (3-L samples) CALIBRATION: solutions of acetone cyanohydrin in ethyl BIAS: not determined acetate OVERALL PRECISION ( ): 0.093 [1] RANGE: 1 to 50 µg per sample ACCURACY: not determined ESTIMATED LOD: 0.1 µg per sample [1] PRECISION ( ): 0.042 [1] APPLICABILITY: The working range is 0.1 to 4.8 ppm (0.33 to 17 mg/m³) for a 3-L air sample. -

Chemical Inventory Packet / Calarp Lists / HFD / Dmg 2004 Table 1
HAYWARD FIRE DEPARTMENT CHEMICAL INVENTORY WORKSHEET To prepare a chemical inventory based on the Hayward Fire Code, you are required to report the quantities of chemicals found in the facility, separated by Hazard Category, and by location. You are also to classify the chemicals as to storage and manner of use. Handling and storage requirements are determined by these factors: the nature of the chemical or its hazard; the location; the manner of use or storage; and the quantity involved in each particular manner of use or storage. A Control Area is a building, a portion of the building, or an outside area where chemicals are allowed to be stored, dispensed, used or handled subject to certain conditions and requirements. This packet consists of the following lists and the Chemical Inventory Worksheet form: 1. Hazardous Materials Hazard Categories. This is the list printed on goldenrod paper. Based on the Uniform Fire Code, this is the classification of chemicals by hazard categories. The different hazard categories are defined and examples are provided for each. Most chemicals exhibit more than one hazard, and all hazards should be considered when preparing your chemical inventory. 2. Tables of Regulated Substances Under the CalARP Program. Printed on pink paper, these are the lists of flammable and toxic substances that, if present at or above certain threshold quantities, will require the preparation of a Risk Management Plan 3. Federal List of Extremely Hazardous Substances (EHS). This list is printed on blue paper. These are the substances that the federal government classifies as extremely hazardous substances. The Hayward Fire Department has included “CalARP-Listed or EHS-Listed Chemicals” as a separate Hazard Category.