N FUEL CHEMISTRY DIVISION ANNUAL PROGRESS REPORT for 1988 S. Vaidyanathan
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ion Sources for High-Power Hadron Accelerators
Ion sources for high-power hadron accelerators Daniel C. Faircloth Rutherford Appleton Laboratory, Chilton, Oxfordshire, UK Abstract Ion sources are a critical component of all particle accelerators. They create the initial beam that is accelerated by the rest of the machine. This paper will introduce the many methods of creating a beam for high-power hadron accelerators. A brief introduction to some of the relevant concepts of plasma physics and beam formation is given. The different types of ion source used in accelerators today are examined. Positive ion sources for producing H+ ions and multiply charged heavy ions are covered. The physical principles involved with negative ion production are outlined and different types of negative ion sources are described. Cutting edge ion source technology and the techniques used to develop sources for the next generation of accelerators are discussed. 1 Introduction 1.1 Ion source basics An ion is an atom or molecule in which the total number of electrons is not equal to the total number of protons, thus giving it a net positive or negative electrical charge. The name ion (from the Greek ιον, meaning "going") was first suggested by William Whewell in 1834. Michael Faraday used the term to refer to the charged particles that carry current in his electrolysis experiments. Ion sources consist of two parts: a plasma generator and an extraction system. The plasma generator must be able to provide enough of the correct ions to the extraction system. There are numerous ways of making plasma: electrical discharges in all of their forms; heating by many different means; using lasers; or even being hit by beams of other particles. -

WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12N 15/10 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/DK20 15/050343 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 11 November 2015 ( 11. 1 1.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: PA 2014 00655 11 November 2014 ( 11. 1 1.2014) DK (84) Designated States (unless otherwise indicated, for every 62/077,933 11 November 2014 ( 11. 11.2014) US kind of regional protection available): ARIPO (BW, GH, 62/202,3 18 7 August 2015 (07.08.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: LUNDORF PEDERSEN MATERIALS APS TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, [DK/DK]; Nordvej 16 B, Himmelev, DK-4000 Roskilde DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (DK). -

Treaty Series
Treaty Series Treaties and internationalagreements registered or filed and recorded with the Secretariat of the United Nations VOLUME 446 Recueil des Traites Traites et accords internationaux enregistres ou classes et inscrits au repertoire au Secrtariat de l'Organisationdes Nations Unies United Nations * Nations Unies New York, 1963 Treaties and international agreements registered or filed and recorded with the Secretariat of the United Nations VOLUME 446 1962 I. Nos. 6397-6409 TABLE OF CONTENTS Treaties and internationalagreements registered /rom 29 November 1962,to 4 December 1962 Page No. 6397. Denmark and People's Republic of China: Exchange of notes constituting an agreement relating to mutual exemption from taxation of residents of either State who are temporarily staying in the other State for educational purposes. Copenhagen, 7 and 23 Septem- ber 1961 .............. .............. ........ 3 No. 6398. United States of America and Denmark: Interim Agreement relating to the General Agreement on Tariffs and Trade (with schedules). Signed at Geneva, on 5 March 1962 ............ 9 No. 6399. United States of America and Finland: Interim Agreement relating to the General Agreement on Tariffs and Trade (with schedules). Signed at Geneva, on 5 March 1962 ......... ... 19 No. 6400. United States of America and Israel: Interim Agreement relating to the General Agreement on Tariffs and Trade (with schedules). Signed at Geneva, on 5 March 1962 ......... ... 29 No. 6401. United States of America and New Zealand: Interim Agreement relating to the General Agreement on Tariffs and Trade (with schedules). Signed at Geneva, on 5 March 1962 ......... ... 39 No. 6402. United States of America and Norway: Interim Agreement relating to the General Agreement on Tariffs and Trade (with schedules). -

WO 2016/196440 Al 8 December 2016 (08.12.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/196440 Al 8 December 2016 (08.12.2016) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61P 3/04 (2006.01) A61K 33/40 (2006.01) kind of national protection available): AE, AG, AL, AM, A61P 9/10 (2006.01) A61K 38/44 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 35/74 (2015.01) A61K 31/17 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/US20 16/034973 KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (22) International Filing Date: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 3 1 May 2016 (3 1.05.2016) PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 62/169,480 1 June 2015 (01 .06.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/327,283 25 April 2016 (25.04.2016) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: XENO BIOSCIENCES INC. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

United States Patents on Powder Metallurgy
UNITED STATES PATENTS ON POWDER METALLURGY UNITED STATES DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS U. S. DEPARTMENT OF COMMERCE W. AVERELL HARRIMAN, Secretary NATIONAL BUREAU OF STANDARDS E. U. CONDON, Director NATIONAL BUREAU OF STANDARDS MISCELLANEOUS PUBLICATION M184 UNITED STATES PATENTS ON POWDER METALLURGY By RAYMOND E. JAGER and ROLLA E. POLLARD Issued July 1 , 1947 UNITED STATES GOVERNMENT PRINTING OFFICE WASHINGTON : 1947 For sale by the Superintendent of Documents, U. S. Government Printing Office Washington 25, D. C. - Price 30 cents PREFACE Patents are disclosures of inventions, in return for which the inventor is given the right to exclude all others from making, using, or selling his invention for the term of 17 years. After this period the invention becomes public property. Patent literature is a valuable source of technical information, for, by these disclosures, the de- velopment of an art may be traced through a long period of time. This publication, based on a collection search of United States patents of the present series, which began in 1836, represents more than a century of progress in the art of powder metallurgy. Patents issued up to January 1, 1947, are included. The collection search was made for the National Bureau of Standards by Invention, Inc., under the direction of Raymond E. Jager, president: R. E Pollard, metallurgist, of the Bureau's staff, edited the abstracts, eliminating those not pertinent to powder metallurgy, and revised the classification. E. U. Condon, Director. n CONTENTS Page. Preface II I. Introduction 1 II. Production of metal powders 1 2 1. Atomization, vaporization, spraying molten metal, or physically contacting it with other fluids, to obtain fine particles 2 2. -
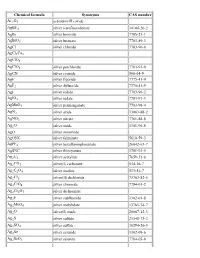
List of Chemical Formulas
Chemical formula Synonyms CAS number Ac2O3 actinium(III) oxide AgBF4 silver tetrafluoroborate 14104-20-2 AgBr silver bromide 7785-23-1 AgBrO3 silver bromate 7783-89-3 AgCl silver chloride 7783-90-6 AgCl3Cu2 AgClO3 AgClO4 silver perchlorate 7783-93-9 AgCN silver cyanide 506-64-9 AgF silver fluoride 7775-41-9 AgF2 silver difluoride 7775-41-9 AgI silver iodide 7783-96-2 AgIO3 silver iodate 7783-97-3 AgMnO4 silver permanganate 7783-98-4 AgN3 silver azide 13863-88-2 AgNO3 silver nitrate 7761-88-8 Ag2O silver oxide 1301-96-8 AgO silver monoxide AgONC silver fulminate 5610-59-3 AgPF6 silver hexafluorophosphate 26042-63-7 AgSNC silver thiocyanate 1701-93-5 Ag2C2 silver acetylide 7659-31-6 Ag2CO3 silver(I) carbonate 534-16-7 Ag2C2O4 silver oxalate 533-51-7 Ag2Cl2 silver(II) dichloride 75763-82-5 Ag2CrO4 silver chromate 7784-01-2 Ag2Cr2O7 silver dichromate Ag2F silver subfluoride 1302-01-8 Ag2MoO4 silver molybdate 13765-74-7 Ag2O silver(I) oxide 20667-12-3 Ag2S silver sulfide 21548-73-2 Ag2SO4 silver sulfate 10294-26-5 Ag2Se silver selenide 1302-09-6 Ag2SeO3 silver selenite 7784-05-6 Ag2SeO4 silver selenate 7784-07-8 Ag2Te silver(I) telluride 12002-99-2 Ag3Br2 silver dibromide 11078-32-3 Ag3Br3 silver tribromide 11078-33-4 Ag3Cl3 silver(III) trichloride 12444-96-1 Ag3I3 silver(III) triiodide 37375-12-5 Ag3PO4 silver phosphate 7784-09-0 AlBO aluminium boron oxide 12041-48-4 AlBO2 aluminium borate 61279-70-7 AlBr aluminium monobromide 22359-97-3 AlBr3 aluminium tribromide 7727-15-3 AlCl aluminium monochloride 13595-81-8 AlClF aluminium chloride fluoride -

Kinetic and Thermodynamic Studies of Reactional System (XIOH) by High Temperature Mass Spectrometry
INSTITUT POLYTECHNIQUE DE GRENOBLE N° attribué par la bibliothèque |__|__|__|__|__|__|__|__|__|__| T H E S E pour obtenir le grade de DOCTEUR DE Grenoble INP Spécialité : « Mécanique des Fluides, Energétique, Procédés » préparée au laboratoire Science et Ingénierie des Matériaux et Procédés dans le cadre de l’Ecole Doctorale « Ingénierie, Matériaux, Mécanique, Environnement, Energétique, Procédés, Production » présentée et soutenue publiquement par Fatima-Zahra ROKI le 29 Janvier 2009 ETUDE DE LA CINETIQUE ET DE LA THERMODYNAMIQUE DES SYSTEMES REACTIONNELS (X-I-O-H) PAR SPECTROMETRIE DE MASSE HAUTE TEMPERATURE tel-00367690, version 1 - 12 Mar 2009 DIRECTEUR DE THESE : Christian Chatillon CO-DIRECTEUR DE THESE : Alexander Pisch JURY M. Yann BULTEL Grenoble INP/LEPMI Président M. Jean-Claude TEDENAC LPMC, Montpellier Rapporteur M. Rudy KONINGS ITU-Euratom, Allemagne Rapporteur M. Christian CHATILLON CNRS / SIMaP, Grenoble Directeur de thèse M. Alexander PISCH CNRS-Lafarge, Lyon Co-Directeur Mme Marie-Noëlle OHNET IRSN / DPAM, Cadarache Examinateur M. Didier JACQUEMAIN IRSN / DS, Cadarache Examinateur tel-00367690, version 1 - 12 Mar 2009 REMERCIEMENTS Ce travail a été réalisé en collaboration avec l’IRSN/DPAM (l’Institut de Radioprotection et Sureté Nucléaire/ Direction de la Prévention des accidents majeurs de Cadarache) que je tiens à remercier de m’avoir choisie en tant que candidate pour la réalisation de ces travaux de recherche. Je remercie, M me Marie-Noëlle OHNET (mon tuteur IRSN), M r Didier Jacquemin, M me Sylvie Fillet et M me Béatrice Simondi-Teisseire pour la confiance, l’autonomie et l’intérêt que vous m’avez accordé tout au long de ce travail. -

Particle Sources
Proceedings of the CERN–Accelerator–School: Introduction to Accelerator Physics Particle Sources D.C. Faircloth Rutherford Appleton Laboratory, Chilton, Oxfordshire, UK Abstract This paper outlines the many ways that the initial beam can be made for particle accelerators. Brief introductions to plasma physics and beam formation are given. Thermionic and photo emission electron guns, with both DC and Radio Frequency (RF) acceleration are outlined. Positive ion sources for producing H+ ions and multiply charged heavy ions are covered. Hot cathode filament sources and cold cathode sources are explored. RF discharge sources (inductively coupled, microwave and ECR) are discussed, as are laser, vacuum arc, and electron beam sources. The physical principles of negative ion production are outlined and different types of negative ion source technologies are described. Polarised particle sources are mentioned briefly. Source choice is summarised and the general practicalities of source operation and development are discussed. 1 Introduction There is a vast zoo of different particle sources out there with many varied applications. Some need to produce ions from tiny samples so they can be analysed and measured. Some need to produce electron beams for imaging, or for welding, or for producing coherent synchrotron light, or for Free Electron Lasers (FEL). Some need to produce ions for industrial processes such as coating, etching, implanting and isotope separation. Some will go into space to provide thrust for satellites. Fusion demands ion sources that generate huge currents of hundreds of amps with beam cross sections measured square meters. Discovery machines demand ever higher beam currents in ever smaller cross sectional areas. -

United States Patent Office Patented
r 2,861,106 United States Patent Office Patented. Nov. 18, 1958 2 rectly, i. e., without prior reduction. In this case, re 2,861,106 duction occurs during the initial phase of the reaction. PROCESS OF PREPARING ALDEHYDES OR KE However, it is advantageous to reduce the catalyst prior TONES BY DEHYDROGENATION OF ALCOHOLS to use in the reaction. Such reduction can be carried Wolfgang Opitz, Knapsack, near Koln, and Werner out by treating the catalyst at relatively low tempera Urbanski, Koln-Bickendorf, Germany, assignors to tures, for instance, between 200° C. and 300° C. with Knapsack - Griesheim Aktiengesellschaft, Knapsack, hydrogen to effect reduction of a considerable amount near Koln, Germany, a corporation of Germany of the cupric oxide to copper and reduction of the high valent chromium to trivalent chromium. If need be, the No Drawing. Application February 24, 1955 O reduction can be carried out by adding nitrogen whereby Serial No. 490,402 too rapid a reaction and a thereby caused sintering can Claims priority, application Germany February 26, 1954 be avoided. A suitable catalyst can, for instance, be prepared as 6 Claims. (CI. 260-596) follows: 5 The present invention relates to a process for the de Cupric oxide is precipitated of its salts by means of hydrogenation of saturated aliphatic alcohols having a sodium carbonate at 80° C. to 90° C., washed until neu straight chain and being of a low molecular weight or tral and dried at 100° C. This fine-grained cupric oxide of cyclic primary and secondary alcohols, preferably al is comminuted and impregnated with an aqueous solu cohols containing 2 to 6 carbon atoms, into the corre 20 tion, for instance a sodium chromate solution of about sponding aldehydes or ketones by leading the alcohol 10% strength, whereby 0.5-3 parts by weight of Cr2O3 vapours, at raised temperatures, over a catalyst compris are calculated for 97-99.5 parts by weight of CuO. -

Roland Friedl Experimental Investigations on the Caesium
Roland Friedl Experimental investigations on the caesium dynamics in H2/D2 low temperature plasmas IPP 4/293 Oktober 2014 AG Experimentelle Plasmaphysik Experimental investigations on the caesium dynamics in H2/D2 low temperature plasmas Dissertation zur Erlangung des Doktorgrades an der Mathematisch-Naturwissenschaftlichen Fakultät der Universität Augsburg vorgelegt von Roland Friedl am 22. August 2013 Vorgelegt am 22. August 2013 Tag der mündlichen Prüfung: 17. Dezember 2013 Erster Gutachter: apl. Prof. Dr.-Ing. U. Fantz Zweiter Gutachter: Prof. Dr. A. Wixforth 3 Abstract The fusion experiment ITER requires powerful neutral beam injection (NBI) systems for heat- ing and current drive. The neutral beam with a power of 16.5 MW at an energy of 1 MeV is generated via accelerating negative hydrogen ions and subsequent neutralization in a gas target. A key component of the NBI system is the ion source which has to provide accelerated current densities of 200 A/m2 D− and 300 A/m2 H−. Such ion sources are currently under development and are based on the surface conversion mechanism: atoms and positive ions from a low temper- ature hydrogen plasma are converted into negative ions at a low work function surface, which is therefore coated with the alkali metal caesium. For that purpose Cs is introduced into the ion source via evaporation from a reservoir. Due to its high chemical reactivity, the adsorbed Cs layer is susceptible to impurities from the residual gas, which degrades the work function of the converter surface. Consequently, the stability and reliability of a high negative ion current density significantly depends on the Cs dynamics in the hydrogen plasma and in the vacuum phases between the pulses. -
Review of Directive 2002/95/EC (Rohs) Categories 8 and 9 - Final Report
Page 1 of 253 RELIABILITY AND FAILURE ANALYSIS Review of Directive 2002/95/EC (RoHS) Categories 8 and 9 - Final Report Dr. Paul Goodman ERA Report 2006-0383 ERA Project 043121538 Final Report Client : European Commission, DG Environment Client Reference : ENV.G.4/ETU/2005/0014 ERA Report Checked by: Approved by: authorised - electronic version authorised - electronic version Dr Paul Goodman Dr Chris Robertson Senior Materials Consultant Head of Reliability and Failure Analysis Reliability and Failure Analysis July 2006 amd. 19 Sep 2006 Ref. 043121538 EC Final report f3.doc 2 ERA Report 2006-0383 - Final Report © Copyright ERA Technology Limited 2006 All Rights Reserved No part of this document may be copied or otherwise reproduced without the prior written permission of ERA Technology Limited. If received electronically, recipient is permitted to make such copies as are necessary to: view the document on a computer system; comply with a reasonable corporate computer data protection and back-up policy and produce one paper copy for personal use. Erratum The original report referred incorrectly on page 11, line 11 and page 248, line 7 to 90/385/EC (the Implanted Medical Devices Directive) whereas this should have been to 98/79/EC (the In-vitro Medical Devices Directive). This has been corrected in the present version. The document may be distributed freely in whole, without alteration, subject to Copyright. ERA Technology Ltd Cleeve Road Leatherhead Surrey KT22 7SA UK Tel : +44 (0) 1372 367444 Fax: +44 (0) 1372 367134 E-mail: [email protected] Read more about ERA Technology on our Internet page at: http://www.era.co.uk/ and about the Reliability and Failure Analysis Group at http://www.era.co.uk/rfa.htm ©Copyright ERA Technology Limited 2006 043121538 EC Final report f3.doc 3 ERA Report 2006-0383 - Final Report Executive Summary Article 6 of 2002/95/EC (RoHS) requires the European Commission to carry out a review of this Directive and present proposals for including Category 8 and 9 equipment within the scope of this Directive.