AIX MARSEILLE UNIVERSITÉ Tam Ngoc PHAM
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ion Sources for High-Power Hadron Accelerators
Ion sources for high-power hadron accelerators Daniel C. Faircloth Rutherford Appleton Laboratory, Chilton, Oxfordshire, UK Abstract Ion sources are a critical component of all particle accelerators. They create the initial beam that is accelerated by the rest of the machine. This paper will introduce the many methods of creating a beam for high-power hadron accelerators. A brief introduction to some of the relevant concepts of plasma physics and beam formation is given. The different types of ion source used in accelerators today are examined. Positive ion sources for producing H+ ions and multiply charged heavy ions are covered. The physical principles involved with negative ion production are outlined and different types of negative ion sources are described. Cutting edge ion source technology and the techniques used to develop sources for the next generation of accelerators are discussed. 1 Introduction 1.1 Ion source basics An ion is an atom or molecule in which the total number of electrons is not equal to the total number of protons, thus giving it a net positive or negative electrical charge. The name ion (from the Greek ιον, meaning "going") was first suggested by William Whewell in 1834. Michael Faraday used the term to refer to the charged particles that carry current in his electrolysis experiments. Ion sources consist of two parts: a plasma generator and an extraction system. The plasma generator must be able to provide enough of the correct ions to the extraction system. There are numerous ways of making plasma: electrical discharges in all of their forms; heating by many different means; using lasers; or even being hit by beams of other particles. -

WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12N 15/10 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/DK20 15/050343 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 11 November 2015 ( 11. 1 1.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: PA 2014 00655 11 November 2014 ( 11. 1 1.2014) DK (84) Designated States (unless otherwise indicated, for every 62/077,933 11 November 2014 ( 11. 11.2014) US kind of regional protection available): ARIPO (BW, GH, 62/202,3 18 7 August 2015 (07.08.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: LUNDORF PEDERSEN MATERIALS APS TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, [DK/DK]; Nordvej 16 B, Himmelev, DK-4000 Roskilde DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (DK). -

Treaty Series
Treaty Series Treaties and internationalagreements registered or filed and recorded with the Secretariat of the United Nations VOLUME 446 Recueil des Traites Traites et accords internationaux enregistres ou classes et inscrits au repertoire au Secrtariat de l'Organisationdes Nations Unies United Nations * Nations Unies New York, 1963 Treaties and international agreements registered or filed and recorded with the Secretariat of the United Nations VOLUME 446 1962 I. Nos. 6397-6409 TABLE OF CONTENTS Treaties and internationalagreements registered /rom 29 November 1962,to 4 December 1962 Page No. 6397. Denmark and People's Republic of China: Exchange of notes constituting an agreement relating to mutual exemption from taxation of residents of either State who are temporarily staying in the other State for educational purposes. Copenhagen, 7 and 23 Septem- ber 1961 .............. .............. ........ 3 No. 6398. United States of America and Denmark: Interim Agreement relating to the General Agreement on Tariffs and Trade (with schedules). Signed at Geneva, on 5 March 1962 ............ 9 No. 6399. United States of America and Finland: Interim Agreement relating to the General Agreement on Tariffs and Trade (with schedules). Signed at Geneva, on 5 March 1962 ......... ... 19 No. 6400. United States of America and Israel: Interim Agreement relating to the General Agreement on Tariffs and Trade (with schedules). Signed at Geneva, on 5 March 1962 ......... ... 29 No. 6401. United States of America and New Zealand: Interim Agreement relating to the General Agreement on Tariffs and Trade (with schedules). Signed at Geneva, on 5 March 1962 ......... ... 39 No. 6402. United States of America and Norway: Interim Agreement relating to the General Agreement on Tariffs and Trade (with schedules). -

WO 2016/196440 Al 8 December 2016 (08.12.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/196440 Al 8 December 2016 (08.12.2016) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61P 3/04 (2006.01) A61K 33/40 (2006.01) kind of national protection available): AE, AG, AL, AM, A61P 9/10 (2006.01) A61K 38/44 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 35/74 (2015.01) A61K 31/17 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/US20 16/034973 KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (22) International Filing Date: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 3 1 May 2016 (3 1.05.2016) PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 62/169,480 1 June 2015 (01 .06.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/327,283 25 April 2016 (25.04.2016) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: XENO BIOSCIENCES INC. -

N FUEL CHEMISTRY DIVISION ANNUAL PROGRESS REPORT for 1988 S. Vaidyanathan
2-o3 BARC/1991/P/002 00 > n o o to FUEL CHEMISTRY DIVISION ANNUAL PROGRESS REPORT FOR 1988 S. Vaidyanathan 1991 GOVERNMENT OF INDIA ATOMIC ENERGY COMMISSION o o U a: < CD FUEL CHEMISTRY DIVISION ANNUAL PROGRESS REPORT FOR 1908 EdiLecl by S. VaidyanaLhan BHADHA ATOMIC RLSLAROI CLN7KI HOMHAY, INDIA 199 I BARC/1991/P/002 BIBLIOGRAPHIC DESCRIPTION SHEET FOR TECHNICAL REPORT (as per IS : 9400 - 1980) 01 Security classification : Uncl assi-f ied 02 Distribution : External 03 Report status : New 04 Series : BARC External 05 Report type : Progress Report 06 Report ND. : BARC/1991/P/002 07 Part No. or Volume No. : 08 Contract No. ; 10 Title and subtitle : Fuel Chemistry Division : annual progress report for 19BB 11 Collation : 158 p., 61 tabs., 13 figs. 13 Project No. : 20 Personal author(s) : S. Vaidyanathan <ed.) 21 Affiliation Df author(s) :Fuel Chemistry Division , Bhabha Atomic Research Centre, Bombay 22 Corporate author(s) : Bhabha Atomic Research Centre, Bombay - 400 0B5 23 Originating unit : Fuel Chemistry Division, BARC, BDHIIJU 24 Sponsor(s) Name ; Department of Atomic Energy Type : Government 30 Date of submission : July 1991 31 Publication/Issue date : August 1991 Contd... <i i) (ii) 40 Publisher/Distributor : Head, Library and Information Division, Bhabha Atomic Research Centre, Bombay 42 Form of distribution : Hard Copy 50 Language of text : Engli sh 51 Language of summary : English 52 No. of references : 53 Gives data on : 60 Abstract : The progress report gives the brief descriptions of various activities of the Fuel Chemistry Division of Bhabha Atomic Research Centre, Bombay for the year 1988. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

United States Patents on Powder Metallurgy
UNITED STATES PATENTS ON POWDER METALLURGY UNITED STATES DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS U. S. DEPARTMENT OF COMMERCE W. AVERELL HARRIMAN, Secretary NATIONAL BUREAU OF STANDARDS E. U. CONDON, Director NATIONAL BUREAU OF STANDARDS MISCELLANEOUS PUBLICATION M184 UNITED STATES PATENTS ON POWDER METALLURGY By RAYMOND E. JAGER and ROLLA E. POLLARD Issued July 1 , 1947 UNITED STATES GOVERNMENT PRINTING OFFICE WASHINGTON : 1947 For sale by the Superintendent of Documents, U. S. Government Printing Office Washington 25, D. C. - Price 30 cents PREFACE Patents are disclosures of inventions, in return for which the inventor is given the right to exclude all others from making, using, or selling his invention for the term of 17 years. After this period the invention becomes public property. Patent literature is a valuable source of technical information, for, by these disclosures, the de- velopment of an art may be traced through a long period of time. This publication, based on a collection search of United States patents of the present series, which began in 1836, represents more than a century of progress in the art of powder metallurgy. Patents issued up to January 1, 1947, are included. The collection search was made for the National Bureau of Standards by Invention, Inc., under the direction of Raymond E. Jager, president: R. E Pollard, metallurgist, of the Bureau's staff, edited the abstracts, eliminating those not pertinent to powder metallurgy, and revised the classification. E. U. Condon, Director. n CONTENTS Page. Preface II I. Introduction 1 II. Production of metal powders 1 2 1. Atomization, vaporization, spraying molten metal, or physically contacting it with other fluids, to obtain fine particles 2 2. -

Case No COMP/M.7393 - ALBEMARLE/ ROCKWOOD
EN Case No COMP/M.7393 - ALBEMARLE/ ROCKWOOD Only the English text is available and authentic. REGULATION (EC) No 139/2004 MERGER PROCEDURE Article 6(1)(b) NON-OPPOSITION Date: 13/11/2014 In electronic form on the EUR-Lex website under document number 32014M7393 Office for Publications of the European Union L-2985 Luxembourg EUROPEAN COMMISSION Brussels, 13.11.2014 C(2014) 8615 final In the published version of this decision, some information has been omitted pursuant to Article 17(2) of Council Regulation (EC) No 139/2004 concerning non-disclosure of business secrets and other confidential information. The omissions are shown thus […]. Where possible the information PUBLIC VERSION omitted has been replaced by ranges of figures or a general description. MERGER PROCEDURE To the notifying party: Dear Sir/Madam, Subject: Case M.7393 – Albemarle/ Rockwood Commission decision pursuant to Article 6(1)(b) of Council Regulation No 139/20041 and article 57 of the EEA agreement (1) On 9 October 2014, the European Commission received notification of a proposed concentration pursuant to Article 4 of the Merger Regulation by which Albemarle Corporation (Albemarle, US) acquires within the meaning of Article 3(1)(b) of the Merger Regulation control of the whole of Rockwood Holdings, Inc. (Rockwood, US) by way of purchase of securities2, hereinafter referred to as "the proposed transaction" or the "Transaction". Albemarle and Rockwood are designated hereinafter as the "Parties". 1. THE PARTIES (2) Albemarle is a global company which develops, manufactures and markets specialty chemicals across a diverse range of end markets including the petroleum refining, consumer electronics, plastics/packaging, construction, automotive, lubricants, pharmaceuticals, crop protection, food safety and custom chemistry services markets. -

Controlling and Exploiting the Caesium Effect in Palladium Catalysed Coupling Reactions
Controlling and exploiting the caesium effect in palladium catalysed coupling reactions Thomas J. Dent Submitted in accordance with the requirements for the degree of Doctor of Philosophy The University of Leeds School of Chemistry May 2019 i The candidate confirms that the work submitted is his own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the report may be published without proper acknowledgement The right of Thomas Dent to be identified as Author of this work has been asserted by him in accordance with the Copyright, Designs and Patents Act 1988. © 2019 The University of Leeds and Thomas J. Dent ii Acknowledgements This project could not have been completed without the help of several individuals who’ve helped guide the project into the finished article. First and foremost I’d like to thank Dr. Bao Nguyen his support, useful discussions and the ability to sift through hundreds of experiments of kinetic data to put together a coherent figure. My writing has come a long way from my transfer report, so all the comments and suggestions seem to have mostly not been in vain. To Paddy, the discussions relating to the NMR studies and anything vaguely inorganic were incredibly useful, and provided me with data that supported our hypothesis with more direct evidence than just the reaction monitoring experiments. Rob, I really enjoyed my time at AZ and your support during my time there was incredibly useful so I could maximise my short secondment when I was getting more results than I knew what to do with. -
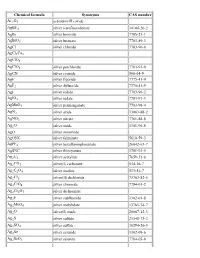
List of Chemical Formulas
Chemical formula Synonyms CAS number Ac2O3 actinium(III) oxide AgBF4 silver tetrafluoroborate 14104-20-2 AgBr silver bromide 7785-23-1 AgBrO3 silver bromate 7783-89-3 AgCl silver chloride 7783-90-6 AgCl3Cu2 AgClO3 AgClO4 silver perchlorate 7783-93-9 AgCN silver cyanide 506-64-9 AgF silver fluoride 7775-41-9 AgF2 silver difluoride 7775-41-9 AgI silver iodide 7783-96-2 AgIO3 silver iodate 7783-97-3 AgMnO4 silver permanganate 7783-98-4 AgN3 silver azide 13863-88-2 AgNO3 silver nitrate 7761-88-8 Ag2O silver oxide 1301-96-8 AgO silver monoxide AgONC silver fulminate 5610-59-3 AgPF6 silver hexafluorophosphate 26042-63-7 AgSNC silver thiocyanate 1701-93-5 Ag2C2 silver acetylide 7659-31-6 Ag2CO3 silver(I) carbonate 534-16-7 Ag2C2O4 silver oxalate 533-51-7 Ag2Cl2 silver(II) dichloride 75763-82-5 Ag2CrO4 silver chromate 7784-01-2 Ag2Cr2O7 silver dichromate Ag2F silver subfluoride 1302-01-8 Ag2MoO4 silver molybdate 13765-74-7 Ag2O silver(I) oxide 20667-12-3 Ag2S silver sulfide 21548-73-2 Ag2SO4 silver sulfate 10294-26-5 Ag2Se silver selenide 1302-09-6 Ag2SeO3 silver selenite 7784-05-6 Ag2SeO4 silver selenate 7784-07-8 Ag2Te silver(I) telluride 12002-99-2 Ag3Br2 silver dibromide 11078-32-3 Ag3Br3 silver tribromide 11078-33-4 Ag3Cl3 silver(III) trichloride 12444-96-1 Ag3I3 silver(III) triiodide 37375-12-5 Ag3PO4 silver phosphate 7784-09-0 AlBO aluminium boron oxide 12041-48-4 AlBO2 aluminium borate 61279-70-7 AlBr aluminium monobromide 22359-97-3 AlBr3 aluminium tribromide 7727-15-3 AlCl aluminium monochloride 13595-81-8 AlClF aluminium chloride fluoride -

“M/S. AJANTA PHARMA LTD.”
APPLICATION FOR ENVIRONMENTAL CLEARANCE OF PROPOSED EXPANSION OF ACTIVE PHARMACEUTICAL INGREDIENTS (API), MANUFACTURING UNIT “M/s. AJANTA PHARMA LTD.” 11KM Stone, Gut No. 378, Plot No 8, Aurangabad –Pune Highway, Village-Waluj, Taluka. Gangapur, District. Aurangabad- 431133. FORM 1 Submitted to Expert Appraisal Committee (Industry-2), MoEFCC, New Delhi Submitted by M/s. AJANTA PHARMA LTD. Environmental Consultant: Building Environment (India) Pvt. Ltd Dakshina Building, Office No. 401, Plot No. 2, Sector-11, CBD Belapur, Navi Mumbai- 400 614 January, 2018. Form 1 for proposed Expansion of API Manufacturing Industry “M/s. Ajanta Pharma Ltd.” at Waluj Village, Taluka-Gangapur, District- Aurangabad, Maharashtra. Form – 1 (I) Basic Information:- Sr. Items Details No. 1. Name of the project Proposed expansion of Active Pharmaceutical Ingredients (API) Manufacturing Industry “M/s. Ajanta Pharma Ltd.” 2. S. No. in the schedule Category 5f as per EIA Notification 2006 & amendments 3. Proposed capacity/ area/ length/ Industry is already engaged in manufacturing 85 nos. of tonnage to be handled/ command API and having production capacity 21.042 MT/Month. area/lease area/ number of wells to In the proposed expansion industry will manufacture 256 be drilled nos. API including existing API. The total production capacity after expansion would remain same as existing i.e. 21.042 MT/Month. Annexure-1 : Details of existing & proposed API 4. New / Expansion / Modernization Expansion. Annexure -2: EC letter of existing unit 5. Existing Capacity/ Area etc. Industry is already engaged in manufacturing 85 nos. of API and having production 21.042 MT/Month. Annexure-3 : Details of existing API 6. -

Kinetic and Thermodynamic Studies of Reactional System (XIOH) by High Temperature Mass Spectrometry
INSTITUT POLYTECHNIQUE DE GRENOBLE N° attribué par la bibliothèque |__|__|__|__|__|__|__|__|__|__| T H E S E pour obtenir le grade de DOCTEUR DE Grenoble INP Spécialité : « Mécanique des Fluides, Energétique, Procédés » préparée au laboratoire Science et Ingénierie des Matériaux et Procédés dans le cadre de l’Ecole Doctorale « Ingénierie, Matériaux, Mécanique, Environnement, Energétique, Procédés, Production » présentée et soutenue publiquement par Fatima-Zahra ROKI le 29 Janvier 2009 ETUDE DE LA CINETIQUE ET DE LA THERMODYNAMIQUE DES SYSTEMES REACTIONNELS (X-I-O-H) PAR SPECTROMETRIE DE MASSE HAUTE TEMPERATURE tel-00367690, version 1 - 12 Mar 2009 DIRECTEUR DE THESE : Christian Chatillon CO-DIRECTEUR DE THESE : Alexander Pisch JURY M. Yann BULTEL Grenoble INP/LEPMI Président M. Jean-Claude TEDENAC LPMC, Montpellier Rapporteur M. Rudy KONINGS ITU-Euratom, Allemagne Rapporteur M. Christian CHATILLON CNRS / SIMaP, Grenoble Directeur de thèse M. Alexander PISCH CNRS-Lafarge, Lyon Co-Directeur Mme Marie-Noëlle OHNET IRSN / DPAM, Cadarache Examinateur M. Didier JACQUEMAIN IRSN / DS, Cadarache Examinateur tel-00367690, version 1 - 12 Mar 2009 REMERCIEMENTS Ce travail a été réalisé en collaboration avec l’IRSN/DPAM (l’Institut de Radioprotection et Sureté Nucléaire/ Direction de la Prévention des accidents majeurs de Cadarache) que je tiens à remercier de m’avoir choisie en tant que candidate pour la réalisation de ces travaux de recherche. Je remercie, M me Marie-Noëlle OHNET (mon tuteur IRSN), M r Didier Jacquemin, M me Sylvie Fillet et M me Béatrice Simondi-Teisseire pour la confiance, l’autonomie et l’intérêt que vous m’avez accordé tout au long de ce travail.