Escherichia Coli HD701
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Centrifan PE Personal Evaporator User Manual
CentrifanTM PE Personal Evaporator User Manual (Shown – CentrifanTM – PE–T; Model 78-0070 with Circulator Coil Accessory) KD Scientific 84 October Hill Road Holliston, MA 01746 Phone: (508) 429-6809 Fax: (508) 893-0160 Part 78-8425 Manual www.kdscientific.com Notices: This system is covered by a limited lifetime warranty. A copy of the warranty is included with this manual. The operator is not required to perform routine maintenance, but procedures for disassembly and cleaning are as described herein. For cleaning procedures, refer to Chapter 4. All information in this manual is subject to change without notice and does not represent a commitment on the part of KD Scientific. The system and various components in the system are the subject of various pending patents. Modular SFC was acquired by Harvard Bioscience in May 2012. KD Scientific is a division of Harvard Bioscience. No part of this manual may be reproduced or transmitted in any form or by any means without the written permission of KD Scientific. All trademarks of other companies are acknowledged to the property of the respective owners. CentrifanTM is a trademark of KD Scientific. Printed in the United States of America. Warnings and Safety Precautions The following precautions should be followed to minimize the possibility of personal injury and/or damage to property while using the CentrifanTM PE. 1. Maintain a Well-Ventilated Laboratory Volatile organic solvents may be used in the system. Ensure that the laboratory is well ventilated so that a buildup of vaporized solvent cannot occur. 2. Ensure That Solvent Vapor Is Not Allowed To Escape Into The Laboratory Any organic solvent vapor must be vented to an external venting system so that it cannot collect in the laboratory. -

Professional and Honor Program - Descriptions with Prerequisite Requirements
Professional and Honor Program - Descriptions with Prerequisite Requirements Professional Career Programs - Full Residence Honor Programs The Professional Career Programs provide broad training for students seeking Short Residence with Self Study Prep rewarding, full-time employment in the recreational diving industry. Both The Institute's Honor programs are shortened versions of our full residence full-residence individual skill and combination skill programs are available. “Professional” Instructor, Divemaster /Dive Control Specialist and Rescue Areas covered in professional programs are: skin and scuba diving instruc- training modules. We designed the “Honor” Programs to give credit for tion, divemaster supervision and dive control specialist, boatmaster, under- certain types of previous training from diver to divemaster and for using our water digital photography and videography with computer editing, promo- self study package to help prepare before attendance. tional diving video-DVD-CD and photo production, detailed dive business, store and resort operations, diving business sales and persuasion, diving Honor program entry requirements are a lot stiffer than our longer full equipment overhaul and repair technology, deep and technical diving residence “Professional” programs. They also do not include the broad instruction, semi-closed or closed circuit rebreather instruction, diving variety of training and practice available in our more extensive "Professional" specialties instruction, submersibles and underwater communication tech- programs. nology, air station and technical gas blending operations, professional CPR- People attending shorter Honor Programs, need to be self-motivated with First Aid, and diving accident response technology and instruction. excellent reading and math skills. Most Honor Programs have physical and Making a good living in the recreational diving industry generally requires written attendance qualification exams to test your preparedness before you more abilities than just teaching. -
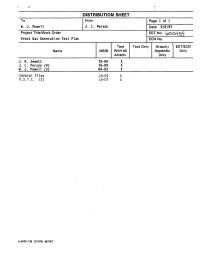
Grout Gas Generation Test Plan ECN No
DISTRIBUTION SHEET To From Page 1 of 1 - W. J. Powell J. C. Person Date 9/8/93 Project Title/Work Order EDTNo. (pOo45^j[ Grout Gas Generation Test Plan ECN No. Text Text Only Attach./ EDT/ECN Name MSIN With All Appendix Only Attach. Only J. R. Jewett T6-09 X J. C. Person (4) T6-09 X W. J. Powell (3) R4-03 X Central Files L8-04 X 0.S.T.I. (2) L8-07 X A-6000-135 (01/93) WEF067 Page 1 of | ai p7 4QQC ENGINEERING DATA TRANSMITTAL I.EDT 600459 2. To: (Receiving Organization) 3. From: (Originating Organization) 4. Related EDT No.: Grout Technology Process Chemistry Laboratory N/A S. Proj./Prog./Dept./Div.: 6. Cog. Engr.: 7. Purchase Order No.: WM/Grout/PAL/FO J. C. Person _N/A_ 8. Originator Remarks: 9. Equip./Component No. N/A 10. Systea/Bldg./Facility: N/A 11. Receiver Remarks: 12. Major Assm. Dwg. No. 13. Permit/Permit Application No.: 14. Required Response Date: 15. DATA TRANSMITTED (F) (G) (H) (I) (A| (C) (D) Reason Origi• Receiv• (E) Title or Description of Data Impact Item Sheet Rev. for nator er (B) Document/Drawing No. Transmitted Level No. No. No. Trans• Dispo• Dispo• mittal sition sition 1. WHCSD-WM-TP-180 Grout Gas Generation 3Q Test Plan 16. KEY Impact Level (F) Reason for Transmittal (G) Disposition (H) & (I) 1, 2, 3, or 4 (see 1. Approval 4. Review 1. Approved 4. Reviewed no/comment MRP 5.431 3Q 2. Release 5. Post-Review 2. Approved w/comment 5. -

A Gas Dilution System for Arsine-Air Mixtures
NBSIR 73-260 A Gas Dilution System for Arsine-Air Mixtures Peter A. Pella, Ernest E. Hughes, and John K. Taylor National Bureau of Standards Department of Commerce Washington, D. C. 20234 October 1973 Final Report Prepared for National Institute for Occupational Safety and Health Division of Laboratories and Criteria Development Cincinnati, Ohio 45202 NBSIR 73-260 A GAS DILUTION SYSTEM FOR ARSINE-AIR MIXTURES Peter A. Pella, Ernest E. Hughes, and John K. Taylor National Bureau of Standards Department of Commerce Washington, D. C. 20234 October 1973 Final Report Prepared for National Institute for Occupational Safety and Health Division of Laboratories and Criteria Development Cincinnati, Ohio 45202 I U. S. DEPARTMENT OF COMMERCE, Frederick B. Dent, Secretary NATIONAL BUREAU OF STANDARDS, Richard W. Roberts, Director A Gas Dilution System for Arsine-Air Mixtures ABSTRACT A gas -blending system originally designed for chlorine-air mixtures was modified for producing arsine-air mixtures in the concentration range from 0.02 to 0.25 ppm. This system has been tested in order to provide accurately known concentrations of arsine- in-air for calibration of analytical monitor- ing devices. An analytical method has been developed for checking the concentration of arsine in the work- ing standard and consists of the spectrophotometric measurement of an arsine-diethyldithiocarbamate complex in solution. 1. INTRODUCTION The system originally designed for preparing chlorine- air mixtures [1] has been adapted for producing arsine-air mixtures in the range from 0.02 to 0.25 ppm. The system combines a gas blending unit and an analytical unit. The gas blending unit produces concentrations of arsine by dilu- tion of a relatively high concentration of arsine in nitrogen (i.e. -

15. Advanced Gas Blender
TDI Standards and Procedures Part 2: TDI Diver Standards 15. Advanced Gas Blender 15.1 Introduction This course enables the successful candidate to engage in the blending of oxygen and helium based gasses. The objective of this course is to train candidates in the proper procedures needed for the preparation and blending of high quality nitrox and trimix gases for use in technical diving. 15.2 Qualifications of Graduates Upon successful completion of this course candidates will be able to prepare high quality scuba gases. 15.3 Who May Teach Any active TDI Advanced Gas Blending Instructor may teach this course 15.4 Student to Instructor Ratio Academic 1. Unlimited, so long as adequate facility, supplies and time are provided to ensure comprehensive and complete training of subject matter Confined Water (swimming pool-like conditions) 1. N/A Open Water (ocean, lake, quarry, spring, river or estuary) 1. N/A 15.5 Student Prerequisites 1. Minimum age 18 2. Provide proof of certification as a TDI Nitrox Gas Blender or equivalent Version 0221 113 TDI Standards and Procedures Part 2: TDI Diver Standards 15.6 Course Structure and Duration Open Water Execution 1. N/A Course Structure 1. TDI allows instructors to structure courses according to the number of students participating and their skill level Duration 1. The minimum number of classroom and briefing hours is 6 15.7 Administrative Requirements The following are the administrative tasks: 1. Collect the course fees from all the students 2. Ensure that the students have the required equipment 3. Communicate the training schedule to the students 4. -

Diving Air Compressor - Wikipedia, the Free Encyclopedia Diving Air Compressor from Wikipedia, the Free Encyclopedia
2/8/2014 Diving air compressor - Wikipedia, the free encyclopedia Diving air compressor From Wikipedia, the free encyclopedia A diving air compressor is a gas compressor that can provide breathing air directly to a surface-supplied diver, or fill diving cylinders with high-pressure air pure enough to be used as a breathing gas. A low pressure diving air compressor usually has a delivery pressure of up to 30 bar, which is regulated to suit the depth of the dive. A high pressure diving compressor has a delivery pressure which is usually over 150 bar, and is commonly between 200 and 300 bar. The pressure is limited by an overpressure valve which may be adjustable. A small stationary high pressure diving air compressor installation Contents 1 Machinery 2 Air purity 3 Pressure 4 Filling heat 5 The bank 6 Gas blending 7 References 8 External links A small scuba filling and blending station supplied by a compressor and Machinery storage bank Diving compressors are generally three- or four-stage-reciprocating air compressors that are lubricated with a high-grade mineral or synthetic compressor oil free of toxic additives (a few use ceramic-lined cylinders with O-rings, not piston rings, requiring no lubrication). Oil-lubricated compressors must only use lubricants specified by the compressor's manufacturer. Special filters are used to clean the air of any residual oil and water(see "Air purity"). Smaller compressors are often splash lubricated - the oil is splashed around in the crankcase by the impact of the crankshaft and connecting A low pressure breathing air rods - but larger compressors are likely to have a pressurized lubrication compressor used for surface supplied using an oil pump which supplies the oil to critical areas through pipes diving at the surface control point and passages in the castings. -

General Training Standards, Policies, and Procedures
General Training Standards, Policies, and Procedures Version 9.2 GUE General Training Standards, Policies, and Procedures © 2021 Global Underwater Explorers This document is the property of Global Underwater Explorers. All rights reserved. Unauthorized use or reproduction in any form is prohibited. The information in this document is distributed on an “As Is” basis without warranty. While every precaution has been taken in its preparation, neither the author(s) nor Global Underwater Explorers have any liability to any person or entity with respect to any loss or damage caused or alleged to be caused, directly or indirectly, by this document’s contents. To report violations, comments, or feedback, contact [email protected]. 2 GUE General Training Standards, Policies, and Procedures Version 9.2 Contents 1. Purpose of GUE .............................................................................................................................................6 1.1 GUE Objectives ............................................................................................................................................. 6 1.1.1 Promote Quality Education .................................................................................................................. 6 1.1.2 Promote Global Conservation Initiatives .......................................................................................... 6 1.1.3 Promote Global Exploration Initiatives ............................................................................................. 6 -

KNF Rotary Evaporator Brochure
RC 900 AND RC 600 ROTARY EVAPORATORS. INSPIRINGLY EASY TO USE. SYSTEMS EXPERTISE BENEFIT FROM EXPERT KNOWLEDGE. ROTARY EVAPORATION TAILORED TO PRACTICAL NEEDS. Under the spotlight at KNF: What aspects are really key What makes KNF’s rotary evaporators stand out? For to rotary evaporation in everyday lab practice? What is years we have offered the SC 920 vacuum pump system, needed to guarantee simple, economical and reliable a much sought-after laboratory device that works side by processes day in day out? These are the questions we side with distilling apparatus. Simple handling, a small used to guide us when developing and implementing the footprint and whisper quiet operation are the KNF RC 900 and the new RC 600. We became involved in daily strengths that set the SC 920 apart. lab work. We asked lab technicians what they wished for, enlisted experts to perform tests, and incorporated their Its success has encouraged us to broaden our systems suggestions. approach to create a perfectly balanced package comprising The result? KNF rotary evaporators designed to impress a rotary evaporator, vacuum pump system, and chiller. thanks to their distinct handling advantages, clever Curious? Then discover KNF’s rotary evaporators and functional details and well thought out safety features. their advantages today! EASY TO USE | CLEVER FUNCTIONAL DETAILS | WELL THOUGHT OUT SAFETY FEATURES RC 900. SUPERIOR PERFORMANCE SYSTEM. RC 600. DUAL EVAPORATION FOR ACADEMIA LABS. Rotary evaporator, vacuum pump system, and chiller as a perfectly System packages to suit different budget conditions are available – coordinated system. e.g., one comprising a vacuum pump system to simultaneously and independently assist two rotary evaporators. -

Case Study: Membrane Co2 Removal from Natural Gas, Grissik Gas Plant, Sumatra, Indonesia
CASE STUDY: MEMBRANE CO2 REMOVAL FROM NATURAL GAS, GRISSIK GAS PLANT, SUMATRA, INDONESIA Charles L. Anderson Air Liquide – MEDAL, L.P. Newport, Delaware 19804 USA (1) 302 225-2102 [email protected] Anggiat Siahaan ConocoPhillips Jakarta, Indonesia (62) (21) 524-1987 [email protected] Abstract One of the world’s largest membrane systems is used for bulk removal of CO2 from natural gas at the Grissik gas processing plant in South Sumatra, Indonesia. The membrane system processes 310 MMSCFD of natural gas, reducing CO2 from 30% to 15%. The Grissik plant is termed a membrane plus amine hybrid which offers particularly attractive operational benefits. The membrane system has been operating for more than four years with no membrane replacement and offers the benefit of very long membrane life. CASE STUDY: MEMBRANE CO2 REMOVAL FROM NATURAL GAS, GRISSIK GAS PLANT, SUMATRA, INDONESIA Charles L. Anderson, Air Liquide – MEDAL, L.P., Newport, Delaware, USA Anggiat Siahaan, ConocoPhillips, Jakarta, Indonesia Introduction ConocoPhillips operates the Grissik gas processing plant on behalf of its partners, Talisman Energy Inc., Pertamina and BPMigas. The Grissik plant, located in South Sumatra, Indonesia, pictured in Figure 1, processes 310 MMSCFD of natural gas, primarily reducing CO2 concentration from 30% in the raw feed down to 3% in the sales gas. Figure 1. Grissik Gas Plant Process Overview The Grissik plant CO2 removal process utilizes both membrane separation and amine absorption technology, termed a membrane plus amine hybrid. A simplified process flow diagram is shown in Figure 2. The Thermal Swing Adsorption (TSA) removes heavy hydrocarbons, serving the triple function of membrane pre-treatment, feed gas dehydration and sales gas hydrocarbon dew pointing. -

TDI Advanced Nitrox Diver
TDI Advanced Nitrox Diver Are you looking to expand your dive time? Maybe you’re a scientific diver or photographer looking to stay in the water a little longer?The TDI Advanced Nitrox Course qualifies divers to use enriched air nitrox from EAN 21 through EAN 100 percent within your current certification level to a maximum depth of 40 metres/130 feet during dives that do not require staged decompression. Often taught in conjunction with the TDI Decompression Procedures course, this can be considered the foundation of your technical diving career. TDI Advanced Nitrox is also a must for SCR or CCR divers. Who this course is for: • The certified nitrox diver looking to expand their understanding of nitrox mixtures containing more than 40% oxygen • The certified nitrox diver looking to expand their in-water skills • The certified diver who has interest in moving forward with technical diving education Course prerequisites (these requirements must be met prior to the start of the course): • Minimum age 18, 15 with parental consent • Minimum certification of TDI Nitrox Diver or equivalent • Proof of 25 logged open water dives What you can expect to learn: Advanced Nitrox picks up where TDI Nitrox leaves off and offers a more in-depth look at diving with nitrox including: • Physics and physiology relating to diving with gas mixes containing more than 40% oxygen • Gas planning, dive tables, dive computers, oxygen limitations, nitrogen limitations • Equipment considerations, cylinder labeling, analyzing nitrox mixtures, gas blending procedures, -

FEI Tecnai 12 ('T12') 120 Kv
Ver. 2017-07-07 Emergency Information: 1. Medical Emergencies: Contact 911 and McGill Security 514.398.3000 2. Leave TEM as is. Do NOT shut down the vacuum system. 3. If possible, turn off High Tension and Close Column Valve. 4. Exit the Room/Building. Emergency Contact Information: • Jeannie Mui – EM Coordinator: Office 514.398.6329; Email: [email protected] • S. Kelly Sears – Facility Manager: Cell 514.576.1926; Email: [email protected] • Joaquin Ortega – Director: Office 514.398.6348; [email protected] FEI Tecnai 12 (‘T12’) 120 kV TEM Simplified Operating Manual (Prepared by S Kelly Sears and Joaquin Ortega) Safety • Do NOT wander behind the microscope or step on cables. • Do NOT touch the sample rod with bare hands below the black O-ring. • There are two computers on the T12 TEM: 1. Microscope PC (MPC) that controls the TEM • Note: Under no circumstances should a user insert a USB device into the MPC. 2. Support PC (SPC) to store and retrieve images (on the desk to the left of the TEM). Protective Equipment • Nitrile gloves for handling sample holder and safety glasses for filling liquid nitrogen dewar. I. Microscope Start Up Note: Your PC desktop may not appear exactly as shown in the images in this manual. a. VERIFY the Red On/Off button and the White Vac. and HT buttons on the right console are lit (currently NOT WORKING). b. LOG IN to the MPC with the username and password created during your training session. After logging in, VERIFY the microscope icon on the right side of the bottom task bar is present and green. -

INNOVATIVE BIOPROCESS EQUIPMENT 2 SOLIDABIOTECH.Com 3
SOLIDABIOTECH.coM INNOVATIVE BIOPROCESS EQUIPMENT 2 SOLIDABIOTECH.coM 3 Solida Biotech is the ideal bioprocess partner to find the solution that suits your specific needs SOLIDA BIOTECH We are a dynamic company that specialises in engineering, developing, and manufacturing in- novative bioprocess equipment. The company was founded by a management team with over 20 years’ experience in the fields of fermentation, cell culture, sterile processing, software and automation technologies, with the aim of meeting the challenges of producing and supplying leading industrial and institutional customers with a new generation of bioreactors and fermenters for research and production, as well as scientific instruments. THE IDEAL BIOPROCESS partner Thanks to the abilities of our skilled team, we can offer flexible and custom made solutions to deal with multiple requirements in the world of green, red and white biotechnologies. Solida offers a wide range of compact and modular bioreactors and fermenters from 50ml to 50 cubic metre pilot and industrial plants and integrated turn-key solutions. All our equipment meets the standards for worldwide distribution. bioprocess solutions We manufacture a wide range of bioreactors, fermenters, at-line analysers, CIP/SIP systems and process vessels for research, development, and production, suitable for fermentation, cell culture, stem cells, re- newable technologies, algae, biomass, biofuel, hydrolysis and more. coMpact bioreactors LaboratorY SIP Bioreactors A compact laboratory bioreactor solution Modular Laboratory SIP stainless steel in characterised by great performance and ease of situ sterilisable bioreactors, from 1 litre up to 50 use, the perfect way to get started. litre w/v as well as custom fit solutions.