Nanopore Sequencing and Hi-C Based De Novo Assembly of Trachidermus Fasciatus Genome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evolutionary Genomics of a Plastic Life History Trait: Galaxias Maculatus Amphidromous and Resident Populations
EVOLUTIONARY GENOMICS OF A PLASTIC LIFE HISTORY TRAIT: GALAXIAS MACULATUS AMPHIDROMOUS AND RESIDENT POPULATIONS by María Lisette Delgado Aquije Submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy at Dalhousie University Halifax, Nova Scotia August 2021 Dalhousie University is located in Mi'kma'ki, the ancestral and unceded territory of the Mi'kmaq. We are all Treaty people. © Copyright by María Lisette Delgado Aquije, 2021 I dedicate this work to my parents, María and José, my brothers JR and Eduardo for their unconditional love and support and for always encouraging me to pursue my dreams, and to my grandparents Victoria, Estela, Jesús, and Pepe whose example of perseverance and hard work allowed me to reach this point. ii TABLE OF CONTENTS LIST OF TABLES ............................................................................................................ vii LIST OF FIGURES ........................................................................................................... ix ABSTRACT ...................................................................................................................... xii LIST OF ABBREVIATION USED ................................................................................ xiii ACKNOWLEDGMENTS ................................................................................................ xv CHAPTER 1. INTRODUCTION ....................................................................................... 1 1.1 Galaxias maculatus .................................................................................................. -

Ecological Effects of the First Dam on Yangtze Main Stream and Future Conservation Recommendations: a Review of the Past 60 Years
Zhang et al.: Ecological effects of the first dam on Yangtze main stream - 2081 - ECOLOGICAL EFFECTS OF THE FIRST DAM ON YANGTZE MAIN STREAM AND FUTURE CONSERVATION RECOMMENDATIONS: A REVIEW OF THE PAST 60 YEARS ZHANG, H.1 – LI, J. Y.1 – WU, J. M.1 – WANG, C. Y.1 – DU, H.1 – WEI, Q. W.1* – KANG, M.2* 1Key Laboratory of Freshwater Biodiversity Conservation, Ministry of Agriculture of China; Yangtze River Fisheries Research Institute, Chinese Academy of Fishery Sciences, Wuhan, Hubei Province, P. R. China (phone: +86-27-8178-0118; fax: +86-27-8178-0118) 2Department of Maritime Police and Production System / The Institute of Marine Industry, Gyeongsang National University, Cheondaegukchi-Gil 38, Tongyeong-si, Gyeongsangnam-do, 53064, South Korea (phone: +82-55-772-9187; fax: +82-55-772-9189) *Corresponding authors e-mail: [email protected]; [email protected] (Received 21st Jul 2017; accepted 27th Oct 2017) Abstract. The Gezhouba Dam was the first and lowermost dam on the major stem of the Yangtze River. Up to now, the dam has been operating for more than 35 years. The time period was a fast economic development stage in the Yangtze basin. Therefore, the entire Yangtze aquatic ecosystem has been highly affected by various anthropogenic activities. Especially, the fish population and distribution in the Yangtze River have been largely altered. This study reviews the ecological effects of the Gezhouba Dam to the Yangtze aquatic biodiversity for the past 60 years based on literatures. It was concluded that the pre-assessment of the Gezhouba Dam on Yangtze fishes in 1970s was appropriate. -

Ioides Species Kazuhiro Takagi, Kunihiko Fujii, Ken-Ichi Yamazaki, Naoki Harada and Akio Iwasaki Download PDF (313.9 KB) View HTML
Aquaculture Journals – Table of Contents With the financial support of Flemish Interuniversity Councel Aquaculture Journals – Table of Contents September 2012 Information of interest !! Animal Feed Science and Technology * Antimicrobial Agents and Chemotherapy Applied and Environmental Microbiology Applied Microbiology and Biotechnology Aqua Aquaculture * Aquaculture Economics & Management Aquacultural Engineering * Aquaculture International * Aquaculture Nutrition * Aquaculture Research * Current Opinion in Microbiology * Diseases of Aquatic Organisms * Fish & Shellfish Immunology * Fisheries Science * Hydrobiologia * Indian Journal of Fisheries International Journal of Aquatic Science Journal of Applied Ichthyology * Journal of Applied Microbiology * Journal of Applied Phycology Journal of Aquaculture Research and Development Journal of Experimental Marine Biology and Ecology * Journal of Fish Biology Journal of Fish Diseases * Journal of Invertebrate Pathology* Journal of Microbial Ecology* Aquaculture Journals Page: 1 of 331 Aquaculture Journals – Table of Contents Journal of Microbiological Methods Journal of Shellfish Research Journal of the World Aquaculture Society Letters in Applied Microbiology * Marine Biology * Marine Biotechnology * Nippon Suisan Gakkaishi Reviews in Aquaculture Trends in Biotechnology * Trends in Microbiology * * full text available Aquaculture Journals Page: 2 of 331 Aquaculture Journals – Table of Contents BibMail Information of Interest - September, 2012 Abstracts of papers presented at the XV International -

Lake Baikal Bibliography, 1989- 1999
UC San Diego Bibliography Title Lake Baikal Bibliography, 1989- 1999 Permalink https://escholarship.org/uc/item/7dc9945d Author Limnological Institute of RAS SB Publication Date 1999-12-31 eScholarship.org Powered by the California Digital Library University of California Lake Baikal Bibliography, 1989- 1999 This is a bibliography of 839 papers published in English in 1989- 1999 by members of Limnological Institute of RAS SB and by their partners within the framework of the Baikal International Center for Ecological Research. Some of the titles are accompanied by abstracts. Coverage is on different aspects of Lake Baikal. Adov F., Takhteev V., Ropstorf P. Mollusks of Baikal-Lena nature reserve (northern Baikal). // World Congress of Malacology: Abstracts; Washington, D.C.: Unitas Malacologica; 1998: 6. Afanasyeva E.L. Life cycle of Epischura baicalensis Sars (Copepoda, Calanoida) in Lake Baikal. // VI International Conference on Copepoda: Abstracts; July 29-August 3, 1996; Oldenburg/Bremerhaven, Germany. Konstanz; 1996: 33. Afanasyeva E.L. Life cycle of Epischura baicalensis Sars (Copepoda, Calanoida) in Lake Baikal. // J. Mar. Syst.; 1998; 15: 351-357. Epischura baicalensis Sars is a dominant pelagic species of Lake Baikal zooplankton. This is endemic to Lake Baikal and inhabits the entire water column. It produces two generations per year: the winter - spring and the summer. These copepods develop under different ecological conditions and vary in the duration of life stages, reproduction time, maturation of sex products and adult males and females lifespan. The total life period of the animals from each generation is one year. One female can produce 10 egg sacks every 10 - 20 days during its life time. -

《中国学术期刊文摘》赠阅 《中国学术期刊文摘》赠阅 CHINESE SCIENCE ABSTRACTS (Monthly, Established in 2006) Vol.10 No.1, 2015 (Sum No.103) Published on January 15, 2015
《中国学术期刊文摘》赠阅 《中国学术期刊文摘》赠阅 CHINESE SCIENCE ABSTRACTS (Monthly, Established in 2006) Vol.10 No.1, 2015 (Sum No.103) Published on January 15, 2015 Chinese Science Abstracts Competent Authority: China Association for Science and Technology Contents Supporting Organization: Department of Society Affairs and Academic Activities China Association for Science and Technology Hot Topic Sponsor: Science and Technology Review Publishing House Ebola Virus………………………………………………………………………1 Publisher: Science and Technology Review Publishing House High Impact Papers Editor-in-chief: CHEN Zhangliang Highly Cited Papers TOP5……………………………………………………11 Chief of the Staff/Deputy Editor-in-chief: Acoustics (11) Agricultural Engineering (12) Agriculture Dairy Animal Science (14) SU Qing [email protected] Agriculture Multidisciplinary (16) Agronomy (18) Allergy (20) Deputy Chief of the Staff/Deputy Editor-in-chief: SHI Yongchao [email protected] Anatomy Morphology (22) Andrology (24) Anesthesiology (26) Deputy Editor-in-chief: SONG Jun Architecture (28) Astronomy Astrophysics (29) Automation Control Systems (31) Deputy Director of Editorial Department: Biochemical Research Methods (33) Biochemistry Molecular Biology (34) WANG Shuaishuai [email protected] WANG Xiaobin Biodiversity Conservation (35) Biology (37) Biophysics (39) Biotechnology Applied Microbiology (41) Cell Biology (42) Executive Editor of this issue: WANG Shuaishuai Cell Tissue Engineering (43) Chemistry Analytical (45) Director of Circulation Department: Chemistry Applied (47) Chemistry Inorganic -
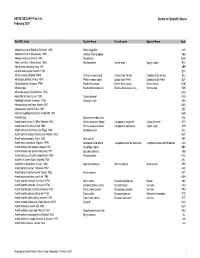
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Genetical Identification and Morphological Description of the Larvae and Juveniles of Porocottus Leptosomus
Shin et al. Fisheries and Aquatic Sciences (2018) 21:37 https://doi.org/10.1186/s41240-018-0115-y RESEARCHARTICLE Open Access Genetical identification and morphological description of the larvae and juveniles of Porocottus leptosomus (Pisces: Cottidae) from Korea Ui Cheol Shin1,3, Yeon Kyu Jeong1, Sang Chul Yoon2, Kwang Ho Choi1 and Jin-Koo Kim3* Abstract The larvae and juveniles of Porocottus leptosomus belonging to the family Cottidae were collected (n = 95, 3.9–16.5 mm in body length, BL) from Busan, Korea, in March 2015. The larvae and juvenile were identified using DNA barcoding as P. leptosomus, and their morphological description was presented in detail. The yolk-sac larvae (3.9–5.6 mm BL) body was slightly compressed, the head was large, the eye was round and large, and the anus was before the middle of the body. The preflexion larvae (5.2–10.0 mm BL) body length drastically increased; caudal fin rays began to occur. The flexion larvae (9.4–11.8 mm BL) notochord flexion started; dorsal, pectoral, and anal fin rays began to occur; pelvic fin buds are seen; they possessed a pair of parietal spine; and a pair of supraocular cirri was first to develop. At 12 mm BL, the notochord was completely flexed. The larva stage (3.9–12.6 mm SL) had the stellate melanophores in the head, isthmus, gut, and tail (along to the ventral midline). During the juvenile stage (11.4–16.5 mm BL), melanophores covered the head and began to form five blackbandsonthesideofthebody.ThelarvaeofP. leptosomus spent pelagic life, but moved to the bottom during the juvenile stage. -

Biology and Ecology of Aqua-Sphere
Chapter II BIOLOGY AND ECOLOGY OF AQUA-SPHERE Toyoji KANEKO, Katsumi TSUKAMOTO, Atsushi TSUDA, Yuzuru SUZUKI and Katsufumi SATOH Irrespective of the nature of the environment, whether it is the sea, river, lake or pond, organisms make even the hot spring and ground water as their habitat. Organisms living in the earth’s aquasphere are called aquatic organisms. Primeval forms of life were born and a major part of history of evolution has progressed in the sea. Therefore, biodiversity in the aquasphere, which has several endemic species, is higher than that in aerosphere and terrasphere. We can find all phyla of prokaryotes, protists, fungi, plants and animals in the aquatic environment. Animals that have strong connection with aquatic environment in their life such as the seabirds and seals are also members of aquatic organisms, and constitute a part of the ecosystem through interrelationships among them. The aquatic environments can be differentiated into riverine, coastal, and deep sea ecosystem. However, as most water bodies on earth including the seas and inland water bodies have some connections to other water bodies, the boundary of the ecosystems is obscure. Ecosystems are connected through migration of organisms and material transportation. In order to live in water, aquatic organisms have developed specialized adaptive functions and organs. The endemic and common organs, tissues and cells such as gills, lateral line, swim-bladder and chloride cells are some examples. These functions characterize not only the behavior of aquatic organisms but also their life history, population structure, reproduction, feeding, migration etc. For example, being denser than air, water offers higher buoyancy to organisms thereby helping large scale dispersion at larval stage, which is a common characteristic trait in aquatic species. -

The First Two Complete Mitochondrial Genomes for the Family Triglidae
www.nature.com/scientificreports OPEN The first two complete mitochondrial genomes for the family Triglidae and implications Received: 20 January 2017 Accepted: 31 March 2017 for the higher phylogeny of Published: xx xx xxxx Scorpaeniformes Lei Cui1, Yuelei Dong1, Fenghua Liu1, Xingchen Gao2, Hua Zhang1, Li Li1, Jingyi Cen1 & Songhui Lu1 The mitochondrial genome (mitogenome) can provide useful information for analyzing phylogeny and molecular evolution. Scorpaeniformes is one of the most diverse teleostean orders and has great commercial importance. To develop mitogenome data for this important group, we determined the complete mitogenomes of two gurnards Chelidonichthys kumu and Lepidotrigla microptera of Triglidae within Scorpaeniformes for the first time. The mitogenomes are 16,495 bp long in C. kumu and 16,610 bp long in L. microptera. Both the mitogenomes contain 13 protein-coding genes (PCGs), 2 ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes and two non-coding regions. All PCGs are initiated by ATG codons, except for the cytochrome coxidase subunit 1 (cox1) gene. All of the tRNA genes could be folded into typical cloverleaf secondary structures, with the exception of tRNASer(AGN) lacks a dihydrouracil (DHU) stem. The control regions are both 838 bp and contain several features common to Scorpaeniformes. The phylogenetic relationships of 33 fish mitogenomes using Bayesian Inference (BI) and Maximum Likelihood (ML) based on nucleotide and amino acid sequences of 13 PCGs indicated that the mitogenome sequences could be useful in resolving higher-level relationship of Scorpaeniformes. The results may provide more insight into the mitogenome evolution of teleostean species. Generally, the fish mitogenome is a circular and double-stranded molecule ranging from 15 to 19 kilobases in length. -

Title Horizontal Distribution and Population Dynamics of The
Horizontal distribution and population dynamics of the Title dominant mysid Hyperacanthomysis longirostris along a temperate macrotidal estuary (Chikugo River estuary, Japan) Author(s) Suzuki, Keita W.; Nakayama, Kouji; Tanaka, Masaru Citation Estuarine, Coastal and Shelf Science (2009), 83(4): 516-528 Issue Date 2009-08 URL http://hdl.handle.net/2433/80150 c 2009 Elsevier Ltd. All rights reserved.; この論文は出版社 版でありません。引用の際には出版社版をご確認ご利用 Right ください。; This is not the published version. Please cite only the published version. Type Journal Article Textversion author Kyoto University Horizontal distribution and population dynamics of the dominant mysid Hyperacanthomysis longirostris along a temperate macrotidal estuary (Chikugo River estuary, Japan) Keita W. Suzukia,*, Kouji Nakayamab, Masaru Tanakab aDivision of Applied Biosciences, Graduate School of Agriculture, Kyoto University, Oiwake-cho, Kitashirakawa, Sakyo-ku, Kyoto 606-8502, Japan bField Science Education and Research Center, Kyoto University, Oiwake-cho, Kitashirakawa, Sakyo-ku, Kyoto 606-8502, Japan *Corresponding author. Tel: 81-75-753-6222. Fax: 81-75-753-6229 E-mail address: [email protected] Postal address: Laboratory of Marine Stock-Enhancement Biology, Division of Applied Biosciences, Graduate School of Agriculture, Kyoto University, Oiwake-cho, Kitashirakawa, Sakyo-ku, Kyoto 606-8502, Japan Abstract The estuarine turbidity maximum (ETM) that develops in the lower salinity areas of macrotidal estuaries has been considered as an important nursery for many fish species. Mysids are one of the dominant organisms in the ETM, serving as a key food source for juvenile fish. To investigate the horizontal distribution and population dynamics of dominant mysids in relation to the fluctuation of physical conditions (temperature, salinity, turbidity, and freshwater discharge), we conducted monthly sampling (hauls of a ring net in the surface water) along the macrotidal Chikugo River estuary in Japan from May 2005 to December 2006. -

Ontogenesis from Embryo to Juvenile and Salinity Tolerance of Japanese
Wang et al. SpringerPlus 2013, 2:289 http://www.springerplus.com/content/2/1/289 a SpringerOpen Journal RESEARCH Open Access Ontogenesis from embryo to juvenile and salinity tolerance of Japanese devil stinger Inimicus japonicus during early life stage Youji Wang†, Lisha Li†, Guoqiang Cui and Weiqun Lu* Abstract Embryonic development and morphological characteristics of Japanese devil stinger Inimicus japonicus during early life stage were investigated. Larvae were hatched out 50 h after fertilization at temperature 21°C. Total length of the newly hatched larva was 4.03 mm, the mouth of the larva opened at 3 days after hatching (DAH), and the yolk sac of the larva disappeared at 5 DAH. After hatching, the pectoral fin first developed, then the tail fin, dorsal fin, anal fin and pelvic fin continuously developed, and all fins formed completely at 15 DAH. The metamorphosis was complete at 25 DAH, and the body color and habit of the metamorphosed individuals were different from the larvae. At 30 DAH, the morphology and habit of the juveniles were the same to adults. In order to determine the suitable salinity for larviculture of I. japonicus, salinity tolerance at different early developmental stages was compared in terms of the survival activity index (SAI) and mean survival time (MST). The results indicated that salinity tolerance varied with development stages. The optimum salinity range for newly hatched larvae was 10– 25‰. Larvae showed low tolerance to low salinity (5‰) before the mouth opened, and the suitable salinities for the larvae with open mouth, yolk-sac larvae, post yolk-sac larvae were 10–15‰. -

University of Southampton Research Repository Eprints Soton
University of Southampton Research Repository ePrints Soton Copyright © and Moral Rights for this thesis are retained by the author and/or other copyright owners. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the copyright holder/s. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the copyright holders. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given e.g. AUTHOR (year of submission) "Full thesis title", University of Southampton, name of the University School or Department, PhD Thesis, pagination http://eprints.soton.ac.uk UNIVERSITY OF SOUTHAMPTON FACULTY OF ENGINEERING AND THE ENVIRONMENT Civil, Maritime and Environmental Engineering and Science Experimental quantification of fish swimming performance and behavioural response to hydraulic stimuli: Application to fish pass design in the UK and China by Lynda Rhian Newbold Thesis for the degree of Doctor of Philosophy February 2015 UNIVERSITY OF SOUTHAMPTON ABSTRACT FACULTY OF ENGINEERING AND THE ENVIRONMENT Civil, Maritime and Environmental Engineering and Science Thesis for the degree of Doctor of Philosophy EXPERIMENTAL QUANTIFICATION OF FISH SWIMMING PERFORMANCE AND BEHAVIOURAL RESPONSE TO HYDRAULIC STIMULI: APPLICATION TO FISH PASS DESIGN IN THE UK AND CHINA Lynda Rhian Newbold Loss of habitat connectivity due to anthropogenic structures is among the greatest threats to freshwater fish populations. Re-establishing river connectivity through fish pass facilities can be an effective and cost-efficient method of enhancing local productivity, yet many are unsuccessful.