Comparative Analyses of Fecal Microbiota in Chinese Isolated Yao
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

EIB-Funded Rare, High-Quality Timber Forest Sustainability Project Non
EIB-funded Rare, High-quality Timber Forest Sustainability Project Non-technical Summary of Environmental Impact Assessment State Forestry Administration December 2013 1 Contents 1、Source of contents ............................... Error! Bookmark not defined. 2、Background information ................................................................... 1 3、Project objectives ................................................................................ 1 4、Project description ............................................................................. 1 4.1 Project site ...................................................................................... 1 4.2 Scope of project .............................................................................. 2 4.3 Project lifecyle .............................................................................. 2 4.4 Alternatives .................................................................................... 3 5、 Factors affecting environment ...................................................... 3 5.1 Positive environmental impacts of the project ............................ 3 5.2 Without-project environment impacts ........................................ 3 5.3 Potential negative envrionmnetal impacts ..................................... 3 5.4 Negative impact mitigation measures ............................................ 4 6、 Environmental monitoring .............................................................. 5 6.1 Environmental monitoring during project implementation .......... -
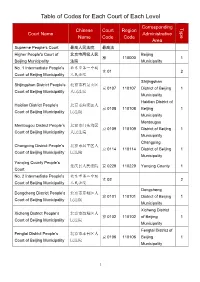
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

Anisotropic Patterns of Liver Cancer Prevalence in Guangxi in Southwest China: Is Local Climate a Contributing Factor?
DOI:http://dx.doi.org/10.7314/APJCP.2015.16.8.3579 Anisotropic Patterns of Liver Cancer Prevalence in Guangxi in Southwest China: Is Local Climate a Contributing Factor? RESEARCH ARTICLE Anisotropic Patterns of Liver Cancer Prevalence in Guangxi in Southwest China: Is Local Climate a Contributing Factor? Wei Deng1&, Long Long2&*, Xian-Yan Tang3, Tian-Ren Huang1, Ji-Lin Li1, Min- Hua Rong1, Ke-Zhi Li1, Hai-Zhou Liu1 Abstract Geographic information system (GIS) technology has useful applications for epidemiology, enabling the detection of spatial patterns of disease dispersion and locating geographic areas at increased risk. In this study, we applied GIS technology to characterize the spatial pattern of mortality due to liver cancer in the autonomous region of Guangxi Zhuang in southwest China. A database with liver cancer mortality data for 1971-1973, 1990-1992, and 2004-2005, including geographic locations and climate conditions, was constructed, and the appropriate associations were investigated. It was found that the regions with the highest mortality rates were central Guangxi with Guigang City at the center, and southwest Guangxi centered in Fusui County. Regions with the lowest mortality rates were eastern Guangxi with Pingnan County at the center, and northern Guangxi centered in Sanjiang and Rongshui counties. Regarding climate conditions, in the 1990s the mortality rate of liver cancer positively correlated with average temperature and average minimum temperature, and negatively correlated with average precipitation. In 2004 through 2005, mortality due to liver cancer positively correlated with the average minimum temperature. Regions of high mortality had lower average humidity and higher average barometric pressure than did regions of low mortality. -

1.9 Alternatives for Dredging
Public Disclosure Authorized ENVIRONMENTAL AND SOCIAL IMPACT ASSESSMENT EXECUTIVE SUMMARY FOR Public Disclosure Authorized HEZHOU URBAN WATER INFRASTRUCTURE AND ENVIRONMENT IMPROVEMENT PROJECT Public Disclosure Authorized Prepared by Guangxi Zhengze Environmental Protection Technology Co., Ltd. Entrusted by Public Disclosure Authorized Hezhou World Bank Loan Project Management Office November 2017 TABLE OF CONTENTS 1. INTRODUCTION .................................................................................................. 1 1.1 Background .................................................................................................. 1 1.2 Environmental Assessment Process and Legal Framework ........................ 1 1.3 Scope of EA and Sensitive Receptors .......................................................... 3 2. PROJECT DESCRIPTION ..................................................................................... 4 3. ENVIRONMENTAL AND SOCIAL BASELINE ................................................. 7 3.1 Physical Environment .................................................................................. 7 3.2 Socio-economic Context.............................................................................. 7 3.3 Ecological Environment .............................................................................. 8 3.4 Environmental Quality ................................................................................ 8 4. ANALYSIS OF ALTERNATIVES ........................................................................ -

Company Board Type Address TPC Country Province/Regio N City
Company Board Type Address TPC Country Province/Regio City n GUANGXI Address:INDUSTRIAL TPC-47 China Guangxi Chongzuo XIANGSHENG PARK, CHENGZHONG HOUSEHOULD TOWN, NINGMING MATERIAL PB COUNTY, CHOUNGZUO TECHNOLOGY CITY, GUANGXI, P.R. CO., LTD CHINA Guangxi MDF Address:Industrial Zone, TPC-47 China Guangxi Qinzhou Dongchang Wood Dasi Town, Qinzhou City, Industry Co., Ltd. China Zhangzhou Thin MDF Address: Shugang TPC-47 China Fujian Zhangzhou Zhongfu New Road,Lingang Industrial Material Co,Ltd. Park, Yunxiao County,Fujian Province Guigang City HWPW Address: The southeastern TPC-47 China Guangxi Guigang Xiangyi Wood of the intersection of Industry Co., Ltd Chengnan Avenue and West Third Road of Guigang Industrial Park(Jiangnan Park),China FOSHAN HWPW Address: Liuchao Shuikou TPC-47 China Guangdong Foshan NANHAI Industrial Zone Lishui Town YINYUAN Nanhai Dist. Foshan City- INSTRUMENTS 528244, Guangdong, China. BOARD MATERIAL MANUFACTURE CO., LTD Fusui Zhongsen HWPW Address:No.5 Fenghua TPC-47 China Guangxi Chongzuo Wood Industry Road, Shanxu Town, Fusui Co., Ltd. County,Chongzuo City, Guangxi Province, China. Guangxi Gaofeng Thin Address:Liutang, Wutang TPC-47 China Guangxi Nanning Wuzhou Wood- MDF&MDF Town, Xingning district, based Panel Co., Nanning City, Guangxi, Ltd. P.R.China Guangxi Fenglin MDF Address:No.26 Mingyang TPC-47 China Guangxi Nanning Wooden-Based Avenue, Wuxu Town, Panels Co., Ltd. Jiangnan District, Nanning City Guangxi MDF Adress:Daqiao TPC-47 China Guangxi Nanning Hengxian Liguan Development Area, Wood-based Hengzhou Town, Heng Panel Co., Ltd. County, Nanning City, Guangxi Guangxi Haolin MDF Address: Qinghu Yashan TPC-47 China Guangxi Nanning Wood-Based town,Bobai County,Yulin Panel Co., Ltd. -

50391-001: Demonstration of Guangxi Elderly Care and Health Care
Resettlement Plan May 2019 People’s Republic of China: Demonstration of Guangxi Elderly Care and Health Care Integration and Public–Private Participation Project Hezhou No. 2 Nursing Home for Disabled Elderly Prepared by Guangxi Zhuang Autonomous Region for the Asian Development Bank. CURRENCY EQUIVALENTS (as of 29 April 2019) Currency unit – yuan (Symbol) CNY1.00 = $6.7297 $1.00 = CNY0.1486 ABBREVIATIONS ADB – Asian Development Bank AFs – affected families APs – affected persons DI – design institute DMS – detailed measurement survey EA – executive agency HLRB – Hezhou Land Resources Administration Bureau HMG – Hezhou Municipal Government HPMO – Hezhou project management office FSR – feasibility study report GDP – gross domestic product GZAR – Guangxi Zhuang Autonomous Region HHs – households IA – implementing agency IMA – independent monitoring agency LA – land acquisition LAR – land acquisition and resettlement LRB – land resources bureau M&E – monitoring and evaluation MLG – minimum living guarantee OP – operation procedures PLG – project leading group PMO – project management office PPTA – project preparation technical assistance PRC – People’s Republic of China RC – residents' committee RIB – resettlement information brochure RO – resettlement office ROW – right-of-way RAP – resettlement plan S&T – science and technology TOR – terms of reference WF – women’s federation km2 – square kilometer mu – A mu is a Chinese unit of measurement (1 mu = 666.667 square meters). NOTE In this report, "$" refers to United States dollars. This resettlement plan is a document of the borrower. The views expressed herein do not necessarily represent those of ADB's Board of Directors, Management, or staff, and may be preliminary in nature. Your attention is directed to the “terms of use” section of this website. -

China 2010 UNGASS Country Progress Report (2008 - 2009)
China 2010 UNGASS Country Progress Report (2008 - 2009) Ministry of Health of the People’s Republic of China 2nd April 2010 1 Table of Contents Table of Contents ............................................................................................................ 2 Status at a glance............................................................................................................ 4 (I) Participation of Stakeholders in Process of Report Drafting................................................4 (II) Overview of China’s AIDS Epidemic.................................................................................5 (III) Policies and Response Work..............................................................................................5 1 Strengthening Leadership. Following Through on Political Commitment....... 6 2 Implementation of the AIDS response........................................................... 7 3 Main Challenges and Solutions....................................................................11 (IV) Overview Table of UNGASS Core Indicator Data .........................................................13 I. Overview of the AIDS epidemic ................................................................................ 21 (I) The rate of increase in the growth of the AIDS epidemic has slowed further....................21 (II) Sexual transmission continues to be the primary mode of transmission, and homosexual transmission is increasing rapidly ...........................................................................................22 -

World Bank Document
The World Bank Hezhou Urban Water Infrastructure and Environment Improvement Project (P158622) Public Disclosure Authorized Public Disclosure Authorized Project Information Document/ Integrated Safeguards Data Sheet (PID/ISDS) Concept Stage | Date Prepared/Updated: 24-Apr-2017 | Report No: PIDISDSC18464 Public Disclosure Authorized Public Disclosure Authorized May 15, 2017 Page 1 of 13 The World Bank Hezhou Urban Water Infrastructure and Environment Improvement Project (P158622) BASIC INFORMATION A. Basic Project Data OPS TABLE Country Project ID Parent Project ID (if any) Project Name China P158622 Hezhou Urban Water Infrastructure and Environment Improvement Project (P158622) Region Estimated Appraisal Date Estimated Board Date Practice Area (Lead) EAST ASIA AND PACIFIC Dec 11, 2017 Mar 15, 2018 Water Financing Instrument Borrower(s) Implementing Agency Investment Project Financing People's Republic of China Hezhou Project Management Office Proposed Development Objective(s) The Project Development Objective (PDO) is to improve flood risk management, reduce water pollution and strengthen water resources planning in Hezhou Municipality. Financing (in USD Million) Finance OLD Financing Source Amount Borrower 150.00 International Bank for Reconstruction and Development 150.00 Total Project Cost 300.00 Environmental Assessment Category Concept Review Decision A-Full Assessment Track I-The review did authorize the preparation to continue May 15, 2017 Page 2 of 13 The World Bank Hezhou Urban Water Infrastructure and Environment Improvement Project (P158622) B. Introduction and Context Country Context 1. China has been urbanizing at very fast rate over the past decades. However, for many medium and small sized cities, investments in the urban infrastructure, especially in the urban water-related assets have not kept pace. -
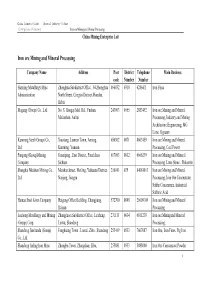
Iron Ore Mining and Mineral Processing China Mining Enterprise Lsit
China Commerce Guide -- Mineral Industry Volume Enterprises Directory Iron ore Mining and Mineral Processing China Mining Enterprise Lsit Iron ore Mining and Mineral Processing Company Name Address Post District Telephone Main Business code Number Number Hanxing Metallurgy Mine Zhonghua Subdistrict Office , 54 Zhonghua 056032 0310 6286411 Iron Fines Administration North Street, Congtai District, Handan, Hebei Magang (Group) Co., Ltd. No. 8, Hongqi Mid. Rd., Yushan, 243003 0555 2883492 Iron ore Mining and Mineral Ma'anshan, Anhui Processing, Industry and Mining Architecture Engineering, MG Lime, Gypsum Kunming Steel (Group) Co., Xiaotang, Lianran Town, Anning, 650302 0871 8603459 Iron ore Mining and Mineral Ltd. Kunming, Yunnan Processing, Coal Power Pangang (Group)Mining Guaziping , East District, Panzhihua, 617063 0812 6666239 Iron ore Mining and Mineral Company Sichuan Processing, Lime Stone,Dolomite Shanghai Meishan Mining Co., Meishan Street, Meiling, Yuhuatai District, 210041 025 84084013 Iron ore Mining and Mineral Ltd. Nanjing, Jiangsu Processing, Iron Ore Concentrate, Sulfur Concentrate, Industrial Sulfuric Acid Hainan Steel & iron Company Haigang Office Building, Changjiang, 572700 0898 26609109 Iron ore Mining and Mineral Hainan Processing Luzhong Metallurgy and Mining Zhangjiawa Subdistrict Office, Laicheng, 271113 0634 6811238 Iron ore Miningand Mineral (Group) Corp. Laiwu, Shandong Processing; Shandong Jinshunda (Group) Fenghuang Town , Lionzi, Zibo , Shandong 255419 0533 7607087 Iron Ore, Iron Fines, Pig Iron Co., Ltd. Shandong -

A Guide to Investment in Hezhou
Positioning A national advanced demonstration zone of circular economy, a new rising industrial city in Guangxi, a regional transportation hub for Guangxi, Guangdong and Hunan, and an outstanding and well-known ecological tourist city in southern China. As “Exploitation of the Works of Nature”, a history book written Name Card in the Ming Dynasty, says "Tin is named He in ancient books, and Linhe (Hezhou) gets its name from the production of tin." Hezhou has a history more than 2,100 years since it was established as Linhe County in 111 B.C., the 6th Yuanding year of Hanwudi in the Western Han National Demonstration Zone of Accepting Industrial Transfer, Chinese National Excellent Tourist City, National Dynasty. In Hejie Town of Babu District, there still stand some Model City of Two-supports, Civilized City of Guangxi, Country of Chinese Hakka People, Longevity Country, Country of well-preserved walls of Nanhan Kingdom built in the period of Five Famous Tea, Country of Rare Stone, and Country of Water Chestnut Dynasties and Ten Kingdoms (907–960 AD). In 226, the 5th Huangwu year of Wu kingdom in the period of Three-kingdoms, Linhe was reestablished as Linhe prefecture. During Environment Overview the period of Sui Dynasty (589–618 AD), Linhe was renamed Hezhou in the 9th Kaihuang year. Located at the place where Guangxi, Guangdong and Hunan provinces meet, Hezhou has been an important business center, since the ancient time, for economic exchanges between Central Plains and the south of the Five Ridges, and for cultural exchange among Hunan, Guangdong, and Guangxi provinces. -

Environmental Impact Assessment Report
E1392 v 1 Certificate of State Environmental Protection Administration Project Serial No. 0502 No. 2902, Grade A Public Disclosure Authorized Guangxi Integrated Forestry Development and Conservation Project Public Disclosure Authorized Environmental Impact Assessment Report Public Disclosure Authorized Environmental Protection Research Institute Public Disclosure Authorized of Guangxi Zhuang Autonomous Region November, 2005 Consigning Unit: Project Management Office of Guangxi Forestry Bureau Assessment Unit: Environmental Protection Research Institute of Guangxi Zhuang Autonomous Region Name of Project: Environmental Impact Assessment on Guangxi Integrated Forestry Development and Conservation Project Legal Representative: Li Min (Director) Examined and Approved by: Zhong Shanjin (Senior Engineer, with a Certificate of Personal Responsibility for Environmental Assessment (CPREA) No. A29020020 ) Verified by: Zeng Guangqing (Senior Engineer, with a CPREA No. A29020015 ) Person in Charge of the Project: Fan Zhenhui (Senior Engineer, with a CPREA No. A29020004 ) Division of Work Table for Report Compilers Name Professiona Number of Educational Period l Title CPREA Contents of Compilation Background of Work and Degree Fan Senior A29020004 Background of the project, Bachelor of 5 months Zhenhui Engineer policies, laws, rules and Environmenta regulations, environmental l Science impact analysis and measures of alleviation, conclusion and suggestions Luo Senior A29020006 General development situation Bachelor of 5 months Yaxue Engineer of natural -

Annual Development Report on China's Trademark Strategy 2013
Annual Development Report on China's Trademark Strategy 2013 TRADEMARK OFFICE/TRADEMARK REVIEW AND ADJUDICATION BOARD OF STATE ADMINISTRATION FOR INDUSTRY AND COMMERCE PEOPLE’S REPUBLIC OF CHINA China Industry & Commerce Press Preface Preface 2013 was a crucial year for comprehensively implementing the conclusions of the 18th CPC National Congress and the second & third plenary session of the 18th CPC Central Committee. Facing the new situation and task of thoroughly reforming and duty transformation, as well as the opportunities and challenges brought by the revised Trademark Law, Trademark staff in AICs at all levels followed the arrangement of SAIC and got new achievements by carrying out trademark strategy and taking innovation on trademark practice, theory and mechanism. ——Trademark examination and review achieved great progress. In 2013, trademark applications increased to 1.8815 million, with a year-on-year growth of 14.15%, reaching a new record in the history and keeping the highest a mount of the world for consecutive 12 years. Under the pressure of trademark examination, Trademark Office and TRAB of SAIC faced the difficuties positively, and made great efforts on soloving problems. Trademark Office and TRAB of SAIC optimized the examination procedure, properly allocated examiners, implemented the mechanism of performance incentive, and carried out the “double-points” management. As a result, the Office examined 1.4246 million trademark applications, 16.09% more than last year. The examination period was maintained within 10 months, and opposition period was shortened to 12 months, which laid a firm foundation for performing the statutory time limit. —— Implementing trademark strategy with a shift to effective use and protection of trademark by law.