A Case of Ezetimibe-Effective Hypercholesterolemia with a Novel Heterozygous Variant in ABCG5
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polymorphisms in APOA1 and LPL Genes Are Statistically Independently Associated with Fasting TG in Men with CAD
European Journal of Human Genetics (2005) 13, 445–451 & 2005 Nature Publishing Group All rights reserved 1018-4813/05 $30.00 www.nature.com/ejhg ARTICLE Polymorphisms in APOA1 and LPL genes are statistically independently associated with fasting TG in men with CAD Olga W Souverein*,1, J Wouter Jukema2, S Matthijs Boekholdt3, Aeilko H Zwinderman1 and Michael WT Tanck1 1Department of Clinical Epidemiology and Biostatistics, Academic Medical Center, Amsterdam, The Netherlands; 2Department of Cardiology, Leiden University Medical Center, Leiden, The Netherlands; 3Department of Cardiology, Academic Medical Center, Amsterdam, The Netherlands The objective of this paper was to identify the single nucleotide polymorphisms (SNPs) that show unshared effects on plasma triglyceride (TG) levels and to investigate whether these SNPs show statistically independent effects on plasma TG levels. In total, 59 polymorphisms in 20 genes involved in lipid metabolism were investigated. Polymorphisms were selected for a multivariate ANOVA model if they showed an univariate association with TG (after adjustment for HDL-C and LDL-C) in more than 50% of bootstrap samples that were made from the original data. The multivariate model included 512 men with coronary artery disease from the REGRESS study who were completely genotyped for eight polymorphisms selected in the univariate procedure (ie, APOA1 G(À75)A, ABCA1 C(À477)T, ABCA1 G1051A, APOC3 T3206G, APOE Arg158Cys, LIPC C(À514)T, LPL Asn291Ser and LPL Ser447Stop). The gene variants APOA1 G(À75)A (P ¼ 0.04) and LPL Asn291Ser (P ¼ 0.03) were significantly associated with plasma TG levels in this multivariate analysis. The eight polymorphisms explained 8.9% of the variation in plasma TG levels. -

The Crucial Roles of Apolipoproteins E and C-III in Apob Lipoprotein Metabolism in Normolipidemia and Hypertriglyceridemia
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Harvard University - DASH The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Sacks, Frank M. 2015. “The Crucial Roles of Apolipoproteins E and C-III in apoB Lipoprotein Metabolism in Normolipidemia and Hypertriglyceridemia.” Current Opinion in Lipidology 26 (1) (February): 56–63. doi:10.1097/mol.0000000000000146. Published Version doi:10.1097/MOL.0000000000000146 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:30203554 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Open Access Policy Articles, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#OAP HHS Public Access Author manuscript Author Manuscript Author ManuscriptCurr Opin Author Manuscript Lipidol. Author Author Manuscript manuscript; available in PMC 2016 February 01. Published in final edited form as: Curr Opin Lipidol. 2015 February ; 26(1): 56–63. doi:10.1097/MOL.0000000000000146. The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia Frank M. Sacks Department of Nutrition, Harvard School of Public Health, Boston, Massachusetts, USA Abstract Purpose of review—To describe the roles of apolipoprotein C-III (apoC-III) and apoE in VLDL and LDL metabolism Recent findings—ApoC-III can block clearance from the circulation of apolipoprotein B (apoB) lipoproteins, whereas apoE mediates their clearance. -

Coexpression of ATP-Binding Cassette Proteins ABCG5 and ABCG8 Permits Their Transport to the Apical Surface
Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface Gregory A. Graf, … , Jonathan C. Cohen, Helen H. Hobbs J Clin Invest. 2002;110(5):659-669. https://doi.org/10.1172/JCI16000. Article Cardiology Mutations in either ATP-binding cassette (ABC) G5 or ABCG8 cause sitosterolemia, an autosomal recessive disorder of sterol trafficking. To determine the site of action of ABCG5 and ABCG8, we expressed recombinant, epitope-tagged mouse ABCG5 and ABCG8 in cultured cells. Both ABCG5 and ABCG8 underwent N-linked glycosylation. When either protein was expressed individually in cells, the N-linked sugars remained sensitive to Endoglycosidase H (Endo H). When ABCG5 and ABCG8 were coexpressed, the attached sugars were Endo H–resistant and neuraminidase-sensitive, indicating that the proteins were transported to the trans-Golgi complex. The mature, glycosylated forms of ABCG5 and ABCG8 coimmunoprecipitated, consistent with heterodimerization of these two proteins. The Endo H–sensitive forms of ABCG5 and ABCG8 were confined to the endoplasmic reticulum (ER), whereas the mature forms were present in non-ER fractions in cultured hepatocytes. Immunoelectron microscopy revealed ABCG5 and ABCG8 on the plasma membrane of these cells. In polarized WIF-B cells, recombinant ABCG5 localized to the apical (canalicular) membrane when coexpressed with ABCG8, but not when expressed alone. To our knowledge this is the first direct demonstration that trafficking of an ABC half-transporter to the cell surface requires the presence of its dimerization partner. Find the latest version: https://jci.me/16000/pdf Coexpression of ATP-binding cassette See the related Commentary beginning on page 605. -

Association Between the APOA2 Rs3813627 Single Nucleotide Polymorphism and HDL and APOA1 Levels Through BMI
biomedicines Article Association between the APOA2 rs3813627 Single Nucleotide Polymorphism and HDL and APOA1 Levels Through BMI Hatim Boughanem 1 , Borja Bandera-Merchán 2, Pablo Hernández-Alonso 2,3,4 , Noelia Moreno-Morales 5, Francisco José Tinahones 2,3, José Lozano 6 , Sonsoles Morcillo 2,3,* and Manuel Macias-Gonzalez 2,3,* 1 Instituto de Investigación Biomédica de Málaga (IBIMA), Facultad de Ciencias, Universidad de Málaga, 29010 Málaga, Spain; [email protected] 2 Unidad de Gestión Clínica de Endocrinología y Nutrición del Hospital Virgen de la Victoria, Instituto de Investigación Biomédica de Málaga (IBIMA), Universidad de Málaga, 29010 Málaga, Spain; [email protected] (B.B.-M.); [email protected] (P.H.-A.); [email protected] (F.J.T.) 3 Centro de Investigación Biomédica en Red de Fisiopatología de la Obesidad y la Nutrición, CIBERObn, 28029 Madrid, Spain 4 Human Nutrition Unit, Faculty of Medicine and Health Sciences, Sant Joan Hospital, Institut d’Investigació Sanitària Pere Virgili, Rovira i Virgili University, 43201 Reus, Spain 5 Department of Physiotherapy, School of Health Sciences, University of Malaga-Instituto de Investigación Biomédica de Málaga (IBIMA), 29010 Málaga, Spain; [email protected] 6 Departamento de Bioquímica y Biología Molecular, Facultad de Ciencias, Universidad de Málaga, 29010 Málaga, Spain; [email protected] * Correspondence: [email protected] (S.M.); [email protected] (M.M.-G.); Tel.: +34-951-032-648 (S.M. & M.M.-G.); Fax: +34-27-951-924-651 (S.M. & M.M.-G.) Received: 18 February 2020; Accepted: 25 February 2020; Published: 27 February 2020 Abstract: Background: The interaction between obesity and genetic traits on high density lipoprotein (HDL) levels has been extensively studied. -

Cardiovascular Disease Products
Cardiovascular Disease Products For more information, visit: www.bosterbio.com Cardiovascular Disease Research Cardiovascular disease is the leading cause of death in developed nations. Boster Bio aims to supply researchers with high-quality antibodies and ELISA kits so they can make new discoveries and help save lives. In this catalogue you will find a comprehensive list of high-affinity Boster antibodies and high sensitivity Boster ELISA kits targeted at proteins associated with cardiovascular disease. Boster: The Fastest Growing About Bosterbio Antibody Company In 2015 Boster is an antibody manufacturer founded in 1993 by histologist Steven Xia. Over the past two decades, Boster and its products have been cited in over 20,000 publications and counting. The firm specializes in developing antibodies and ELISA kits that feature high affinity, Boster Bio received the CitaAb award for high specificity at affordable the greatest increase in number of prices. citations during 2015 than any other antibody manufacturer. Table of Contents Boster Cardiovascular Disease Related Antibodies…………..………..... 2 Boster Cardiovascular Disease Related ELISA Kits……………………..…. 9 1 High Affinity Boster Antibodies Boster supplies only the highest quality antibodies. Our high-affinity polyclonal and monoclonal antibodies are thoroughly validated by Western Blotting, Immunohistochemistry and ELISA. This is our comprehensive catalog of our antibody products related to cardiovascular disease, sorted in alphabetical order by target gene name. Catalog No Product Name -
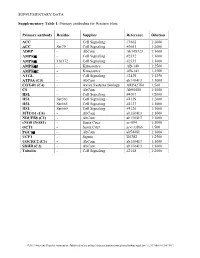
Supplementary Table 1
SUPPLEMENTARY DATA Supplementary Table 1. Primary antibodies for Western blots. Primary antibody Residue Supplier Reference Dilution ACC - Cell Signaling #3662 1:2000 ACC Ser79 Cell Signaling #3661 1:2000 ADRP - AbCam Ab108323 1:1000 AMPKα - Cell Signaling #2532 1:1000 AMPKα Thr172 Cell Signaling #2535 1:1000 AMPKα1 - Kinasource AB-140 1:2500 AMPKα2 - Kinasource AB-141 1:2500 ATGL - Cell Signaling #2439 1:1250 ATP5A (C5) - AbCam ab110413 1:1000 COX4l1 (C4) - Aviva Systems Biology ARP42784 1:500 CS - AbCam Ab96600 1:1000 HSL - Cell Signaling #4107 1:2000 HSL Ser563 Cell Signaling #4139 1:2000 HSL Ser565 Cell Signaling #4137 1:1000 HSL Ser660 Cell Signaling #4126 1:1000 MTCO1 (C4) - AbCam ab110413 1:1000 NDUFB8 (C1) - AbCam ab110413 1:1000 eNOS (NOS3) - Santa Cruz sc-654 1:1000 OCT1 - Santa Cruz sc-133866 1:500 PGC1α - AbCam ab54481 1:1000 UCP1 - Sigma U6382 1:2500 UQCRC2 (C3) - AbCam ab110413 1:1000 SDHB (C2) - AbCam ab110413 1:1000 Tubulin - Cell Signaling #2148 1:2000 ©2013 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0194/-/DC1 SUPPLEMENTARY DATA Supplementary Table 2. Primer sequences for qRT-PCR. Gene Accession number Forward primer Reverse primer Abca1 NM_013454.3 CCCAGAGCAAAAAGCGACTC GGTCATCATCACTTTGGTCCTTG Abcg5 NM_031884 TGTCCTACAGCGTCAGCAACC GGCCACTCTCGATGTACAAGG Abcg8 NM_026180 TCCTGTGAGCTGGGCATCCGA CCCGCAGCCTGAGCTCCCTAT Acaca NM_133360.2 CAGCTGGTGCAGAGGTACCG TCTACTCGCAGGTACTGCCG Acacb NM_133904.2 GCGCCTACTATGAGGCCCAGCA ACAAACTCGGCTGGGGACGC Acly NM_134037.2 -

Overexpression of ABCG5 and ABCG8 Promotes Biliary Cholesterol Secretion and Reduces Fractional Absorption of Dietary Cholestero
Overexpression of ABCG5 See the related Commentary beginning on page 605. and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol Liqing Yu,1 Jia Li-Hawkins,1 Robert E. Hammer,2,3 Knut E. Berge,1 Jay D. Horton,1 Jonathan C. Cohen,4 and Helen H. Hobbs1 1Department of Molecular Genetics and McDermott Center for Human Growth and Development, 2Department of Biochemistry, 3Howard Hughes Medical Institute, and 4Center for Human Nutrition, University of Texas Southwestern Medical Center at Dallas, Dallas, Texas, USA Two ATP-binding cassette (ABC) transporters, ABCG5 and ABCG8, have been proposed to limit sterol absorption and to promote biliary sterol excretion in humans. To test this hypothesis, a P1 clone containing the human ABCG5 and ABCG8 genes was used to generate transgenic mice. The transgenes were expressed primarily in the liver and small intestine, mirroring the expression pattern of the endogenous genes. Transgene expression only modestly affected plasma and liver cholesterol levels but profoundly altered cholesterol transport. The fractional absorption of dietary cholesterol was reduced by about 50%, and biliary cholesterol levels were increased more than fivefold. Fecal neu- tral sterol excretion was increased three- to sixfold and hepatic cholesterol synthesis increased two- to fourfold in the transgenic mice. No significant changes in the pool size, composition, and fecal excretion of bile acids were observed in the transgenic mice. Transgene expression attenuated the increase in hepatic cholesterol content induced by consumption of a high cholesterol diet. These results demonstrate that increased expression of ABCG5 and ABCG8 selectively drives biliary neutral sterol secretion and reduces intestinal cholesterol absorption, leading to a selective increase in neu- tral sterol excretion and a compensatory increase in cholesterol synthesis. -

Low-Density Lipoprotein Receptor–Dependent and Low-Density Lipoprotein Receptor–Independent Mechanisms of Cyclosporin A–Induced Dyslipidemia
Original Research Low-Density Lipoprotein Receptor–Dependent and Low-Density Lipoprotein Receptor–Independent Mechanisms of Cyclosporin A–Induced Dyslipidemia Maaike Kockx, Elias Glaros, Betty Kan, Theodore W. Ng, Jimmy F.P. Berbée, Virginie Deswaerte, Diana Nawara, Carmel Quinn, Kerry-Anne Rye, Wendy Jessup, Patrick C.N. Rensen, Peter J. Meikle, Leonard Kritharides Objective—Cyclosporin A (CsA) is an immunosuppressant commonly used to prevent organ rejection but is associated with hyperlipidemia and an increased risk of cardiovascular disease. Although studies suggest that CsA-induced hyperlipidemia is mediated by inhibition of low-density lipoprotein receptor (LDLr)–mediated lipoprotein clearance, the data supporting this are inconclusive. We therefore sought to investigate the role of the LDLr in CsA-induced hyperlipidemia by using Ldlr-knockout mice (Ldlr−/−). Approach and Results—Ldlr−/− and wild-type (wt) C57Bl/6 mice were treated with 20 mg/kg per d CsA for 4 weeks. On a chow diet, CsA caused marked dyslipidemia in Ldlr−/− but not in wt mice. Hyperlipidemia was characterized by a prominent increase in plasma very low–density lipoprotein and intermediate-density lipoprotein/LDL with unchanged plasma high-density lipoprotein levels, thus mimicking the dyslipidemic profile observed in humans. Analysis of specific lipid species by liquid chromatography–tandem mass spectrometry suggested a predominant effect of CsA on increased very low–density lipoprotein–IDL/LDL lipoprotein number rather than composition. Mechanistic studies indicated that CsA did not alter hepatic lipoprotein production but did inhibit plasma clearance and hepatic uptake of [14C]cholesteryl oleate and glycerol tri[3H]oleate-double-labeled very low–density lipoprotein–like particles. -

Common Genetic Variations Involved in the Inter-Individual Variability Of
nutrients Review Common Genetic Variations Involved in the Inter-Individual Variability of Circulating Cholesterol Concentrations in Response to Diets: A Narrative Review of Recent Evidence Mohammad M. H. Abdullah 1 , Itzel Vazquez-Vidal 2, David J. Baer 3, James D. House 4 , Peter J. H. Jones 5 and Charles Desmarchelier 6,* 1 Department of Food Science and Nutrition, Kuwait University, Kuwait City 10002, Kuwait; [email protected] 2 Richardson Centre for Functional Foods & Nutraceuticals, University of Manitoba, Winnipeg, MB R3T 6C5, Canada; [email protected] 3 United States Department of Agriculture, Agricultural Research Service, Beltsville, MD 20705, USA; [email protected] 4 Department of Food and Human Nutritional Sciences, University of Manitoba, Winnipeg, MB R3T 2N2, Canada; [email protected] 5 Nutritional Fundamentals for Health, Vaudreuil-Dorion, QC J7V 5V5, Canada; [email protected] 6 Aix Marseille University, INRAE, INSERM, C2VN, 13005 Marseille, France * Correspondence: [email protected] Abstract: The number of nutrigenetic studies dedicated to the identification of single nucleotide Citation: Abdullah, M.M.H.; polymorphisms (SNPs) modulating blood lipid profiles in response to dietary interventions has Vazquez-Vidal, I.; Baer, D.J.; House, increased considerably over the last decade. However, the robustness of the evidence-based sci- J.D.; Jones, P.J.H.; Desmarchelier, C. ence supporting the area remains to be evaluated. The objective of this review was to present Common Genetic Variations Involved recent findings concerning the effects of interactions between SNPs in genes involved in cholesterol in the Inter-Individual Variability of metabolism and transport, and dietary intakes or interventions on circulating cholesterol concen- Circulating Cholesterol trations, which are causally involved in cardiovascular diseases and established biomarkers of Concentrations in Response to Diets: cardiovascular health. -

Supplemental Data
Supplemental Table 1. Panels (utilized in this study) and the genes included in each of them are summarized below. Nephrotic Syndrome (NS)/Focal ACTN4, ANKFY1, ANLN, APOL1, ARHGAP24, ARHGDIA, CD2AP, CDK20, COL4A3, COL4A4, COL4A5, COL4A6, COQ2, COQ6, COQ8B, CRB2, CUBN, Segmental DGKE, DLC1, EMP2, FAT1, GAPVD1, GON7, INF2, ITGA3, ITGB4, ITSN1, ITSN2, KANK1, KANK2, KANK4, KAT2B, KIRREL1, LAGE3, LAMA5, LAMB2, Glomerulosclerosis LMX1B, MAFB, MAGI2, MYH9, MYO1E, NEU1, NFKB2, NPHS1, NPHS2, NUP107, NUP133, NUP160, NUP205, NUP93, OSGEP, PAX2, PDSS2, PLCE1, (FSGS) Panel PTPRO, SCARB2, SGPL1, SMARCAL1, TBC1D8B, TNS2, TP53RK, TPRKB, TRIM8, TRPC6, TTC21B, WDR4, WDR73, WT1, XPO5, YRDC Alport syndrome COL4A3, COL4A4, COL4A5, COL4A6 Panel Autosomal Dominant Polycystic Kidney DNAJB11, GANAB, HNF1B, PKD1, PKD2 Disease (ADPKD) panel Recessive Polycystic Kidney DZIP1L, PKHD1 Disease (ARPKD) panel Hereditary cystic ANKS6, CEP164, CEP290, CEP83, COL4A1, CRB2, DCDC2, DICER1, DNAJB11, DZIP1L, GANAB, GLIS2, HNF1B, IFT172, INVS, IQCB1, JAG1, LRP5, kidney disease MAPKBP1, MUC1, NEK8, NOTCH2, NPHP1, NPHP3, NPHP4, OFD1, PAX2, PKD1, PKD2, PKHD1, RPGRIP1L, SDCCAG8, SEC61A1, TMEM67, TSC1, panel TSC2, TTC21B, UMOD, VHL, WDR19, ZNF423 Nephrotic NPHS1, NPHS2, WT1, PLCE1, LAMB2 Syndrome Autosomal Dominant and Recessive Polycystic Kidney DNAJB11, DZIP1L, GANAB, HNF1B, PKD1, PKD2, PKHD1 Disease (ADPKD and ARPKD) Panel Distal Renal Tubular Acidosis ATP6V0A4, ATP6V1B1, CA2, SLC4A1 Panel Atypical Hemolytic Uremic syndrome C3, CFB, CFH, CFHR1, CFHR3, CFHR5, CFI, DGKE, MCP, THBD -

Cholesterol and Saturated Fat Intake Determine the Effect Of
article September 2006 ⅐ Vol. 8 ⅐ No. 9 Cholesterol and saturated fat intake determine the effect of polymorphisms at ABCG5/ABCG8 genes on lipid levels in children Enrique Viturro, PhD1, Manuel de Oya, PhD, MD1, Miguel A. Lasuncio´n, PhD2, Lydia Gorgojo, PhD, MD3, Jose´ M. Martı´n Moreno, PhD, MD3, Mercedes Benavente, PhD1, Beatriz Cano, PhD1, and Carmen Garces, PhD1 Purpose: Analysis of mutations in genes of the cholesterol metabolic pathway has not completely explained the interindividual variability of blood cholesterol concentrations attributed to gene–nutrient interactions. Thus, we analyzed polymorphisms in the ABCG5 and ABCG8 genes, involved in the regulation of intestinal cholesterol absorption, with special interest in a potential interaction with diet to determine lipid levels. Methods: The polymorphisms ABCG5 C1950G (Gln604Glu) and ABCG8 C1895T (Ala640Val) were determined by polymerase chain reaction and restriction analysis in 1227 healthy school children, aged 6 to 8 years. Results: No significant differences were found in blood lipid levels between subjects with different genotypes of the two analyzed polymorphisms. However, important differences appeared when separating subjects by their different lipid intake. The presence of the ABCG8 C1895T and ABCG5 C1950G polymorphisms was associated with different plasma total cholesterol, low-density lipoprotein cholesterol complex, and apolipoprotein B levels only in low-cholesterol consumers (significantly for the C1895T polymorphism), and among children within the lower tertile of saturated fat intake (significantly for the C1950G polymorphism). Conclusion: Polymorphisms at the half-transporter ABCG5 and ABCG8 genes affect blood cholesterol concentrations in prepubertal children by influencing dietary respon- siveness. This highly significant gene–nutrient interaction could explain the great individual differences in the plasma lipid response to cholesterol and fat intake. -

Progress in Lipid Research Progress in Lipid Research 46 (2007) 297–314 Review Non-Vesicular Sterol Transport in Cells
Progress in Lipid Research Progress in Lipid Research 46 (2007) 297–314 www.elsevier.com/locate/plipres Review Non-vesicular sterol transport in cells William A. Prinz * Laboratory of Cell Biochemistry and Biology, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, US Department of Health and Human Services, Bethesda, MD 20892, USA Abstract Sterols such as cholesterol are important components of cellular membranes. They are not uniformly distributed among organelles and maintaining the proper distribution of sterols is critical for many cellular functions. Both vesicular and non- vesicular pathways move sterols between membranes and into and out of cells. There is growing evidence that a number of non-vesicular transport pathways operate in cells and, in the past few years, a number of proteins have been proposed to facilitate this transfer. Some are soluble sterol transfer proteins that may move sterol between membranes. Others are inte- gral membranes proteins that mediate sterol efflux, uptake from cells, and perhaps intracellular sterol transfer as well. In most cases, the mechanisms and regulation of these proteins remains poorly understood. This review summarizes our cur- rent knowledge of these proteins and how they could contribute to intracellular sterol trafficking and distribution. Published by Elsevier Ltd. Keywords: Cholestrol; Transport; Non-vesicular; Membranes; Lipid transport proteins Contents 1. Introduction ...................................................................... 298 2. Non-vesicular sterol transport pathways .................................................. 299 2.1. Transfer of cholesterol to the outer and inner mitochondrial membranes . .................... 300 2.2. PM to ERC sterol transport . ........................................................ 300 2.3. Sterol movement to and from lipid droplets (LDs). ........................................ 300 2.4.