Human Apoa-I Overexpression Enhances Macrophage-Specific Reverse Cholesterol Transport but Fails to Prevent Inherited Diabesity in Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Heterozygous ATP-Binding Cassette Transporter G5 Gene Deficiency and Risk of Coronary Artery Disease
bioRxiv preprint doi: https://doi.org/10.1101/780734; this version posted September 27, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Heterozygous ATP-binding Cassette Transporter G5 Gene Deficiency and Risk of Coronary Artery Disease Short title: Heterozygous ABCG5 deficiency and risk of CAD Akihiro Nomura*, MD PhD, Connor A. Emdin*, DPhil, Hong Hee Won, PhD, Gina M. Peloso, PhD, Pradeep Natarajan, MD, Diego Ardissino, MD, John Danesh, FRCP DPhil, Heribert Schunkert, MD, Adolfo Correa, MD PhD, Matthew J. Bown, MD FRCS, Nilesh J. Samani, MD FRCP, Jeanette Erdmann, PhD, Ruth McPherson, MD, Hugh Watkins, MD PhD, Danish Saleheen, MD, Roberto Elosua, MD PhD, Masa-aki Kawashiri, MD PhD, Hayato Tada, MD PhD, Namrata Gupta, PhD, Svati H. Shah, MD MHS, Daniel J. Rader, MD, Stacey Gabriel, PhD, Amit V. Khera*, MD, Sekar Kathiresan*, MD *: These authors contributed equally Address for correspondence: Sekar Kathiresan, MD Verve Therapeutics 26 Landsdowne Street, 1st Floor Cambridge, MA 02139 Email: [email protected] Phone: 617 603 0070 bioRxiv preprint doi: https://doi.org/10.1101/780734; this version posted September 27, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Background: Familial sitosterolemia is a rare, recessive Mendelian disorder characterized by hyperabsorption and decreased biliary excretion of dietary sterols. -

The Effect of Statin Treatment on Intratumoral Cholesterol Levels and LDL Receptor Expression: a Window-Of-Opportunity Breast Ca
Feldt et al. Cancer & Metabolism (2020) 8:25 https://doi.org/10.1186/s40170-020-00231-8 RESEARCH Open Access The effect of statin treatment on intratumoral cholesterol levels and LDL receptor expression: a window-of- opportunity breast cancer trial Maria Feldt1,2* , Julien Menard1, Ann H. Rosendahl1,2, Barbara Lettiero1, Pär-Ola Bendahl1, Mattias Belting1,2,3 and Signe Borgquist1,4 Abstract Background: Deregulated lipid metabolism is common in cancer cells and the mevalonate pathway, which synthesizes cholesterol, is central in lipid metabolism. This study aimed to assess statin-induced changes of the intratumoral levels of cholesterol and the expression of the low-density lipoprotein receptor (LDLR) to enhance our understanding of the role of the mevalonate pathway in cancer cholesterol metabolism. Methods: This study is based on a phase II clinical trial designed as a window-of-opportunity trial including 50 breast cancer patients treated with 80 mg of atorvastatin/day for 2 weeks, between the time of diagnosis and breast surgery. Lipids were extracted from frozen tumor tissue sampled pre- and post-atorvastatin treatment. Intratumoral cholesterol levels were measured using a fluorometric quantitation assay. LDLR expression was evaluated by immunohistochemistry on formalin-fixed paraffin-embedded tumor tissue. Paired blood samples pre- and post- atorvastatin were analyzed for circulating low-density lipoprotein (LDL), high-density lipoprotein (HDL), apolipoprotein A1, and apolipoprotein B. In vitro experiments on MCF-7 breast cancer cells treated with atorvastatin were performed for comparison on the cellular level. Results: In the trial, 42 patients completed all study parts. From the paired tumor tissue samples, assessment of the cholesterol levels was achievable for 14 tumors, and for the LDLR expression in 24 tumors. -

Participation of ABCA1 Transporter in Pathogenesis of Chronic Obstructive Pulmonary Disease
International Journal of Molecular Sciences Review Participation of ABCA1 Transporter in Pathogenesis of Chronic Obstructive Pulmonary Disease Stanislav Kotlyarov Department of Nursing, Ryazan State Medical University, 390026 Ryazan, Russia; [email protected] Abstract: Chronic obstructive pulmonary disease (COPD) is the important medical and social problem. According to modern concepts, COPD is a chronic inflammatory disease, macrophages play a key role in its pathogenesis. Macrophages are heterogeneous in their functions, which is largely determined by their immunometabolic profile, as well as the features of lipid homeostasis, in which the ATP binding cassette transporter A1 (ABCA1) plays an essential role. The objective of this work is the analysis of the ABCA1 protein participation and the function of reverse cholesterol transport in the pathogenesis of COPD. The expression of the ABCA1 gene in lung tissues takes the second place after the liver, which indicates the important role of the carrier in lung function. The participation of the transporter in the development of COPD consists in provision of lipid metabolism, regulation of inflammation, phagocytosis, and apoptosis. Violation of the processes in which ABCA1 is involved may be a part of the pathophysiological mechanisms, leading to the formation of a heterogeneous clinical course of the disease. Keywords: chronic obstructive pulmonary disease; COPD; inflammation; ABCA1; reverse cholesterol transport; innate immune system Citation: Kotlyarov, S. Participation of ABCA1 Transporter in Pathogenesis of Chronic Obstructive 1. Introduction Pulmonary Disease. Int. J. Mol. Sci. Chronic obstructive pulmonary disease (COPD) is one of the most widespread dis- 2021, 22, 3334. https://doi.org/ eases, it has great medical significance due to the high frequency of temporary and per- 10.3390/ijms22073334 sistent disability and mortality. -

1 CETP Inhibition Improves HDL Function but Leads to Fatty Liver and Insulin Resistance in CETP-Expressing Transgenic Mice on A
Page 1 of 55 Diabetes CETP inhibition improves HDL function but leads to fatty liver and insulin resistance in CETP-expressing transgenic mice on a high-fat diet Lin Zhu1,2, Thao Luu2, Christopher H. Emfinger1,2, Bryan A Parks5, Jeanne Shi2,7, Elijah Trefts3, Fenghua Zeng4, Zsuzsanna Kuklenyik5, Raymond C. Harris4, David H. Wasserman3, Sergio Fazio6 and John M. Stafford1,2,3,* 1VA Tennessee Valley Healthcare System, 2Division of Diabetes, Endocrinology, & Metabolism, 3Department of Molecular Physiology and Biophysics, 4Devision of Nephrology and Hypertension, Vanderbilt University School of Medicine. 5Division of Laboratory Sciences, Centers for Disease Control and Prevention. 6The Center for Preventive Cardiology at the Knight Cardiovascular Institute, Oregon Health & Science University. 7Trinity College of Art and Science, Duke University. * Address correspondence and request for reprints to: John. M. Stafford, 7445D Medical Research Building IV, Nashville, TN 37232-0475, phone (615) 936-6113, fax (615) 936- 1667 Email: [email protected] Running Title: CETP inhibition and insulin resistance Word Count: 5439 Figures: 7 Tables: 1 1 Diabetes Publish Ahead of Print, published online September 13, 2018 Diabetes Page 2 of 55 Abstract In clinical trials inhibition of cholesteryl ester transfer protein (CETP) raises HDL cholesterol levels but doesn’t robustly improve cardiovascular outcomes. About 2/3 of trial participants were obese. Lower plasma CETP activity is associated with increased cardiovascular risk in human studies, and protective aspects of CETP have been observed in mice fed a high-fat diet (HFD) with regard to metabolic outcomes. To define if CETP inhibition has different effects depending on the presence of obesity, we performed short- term anacetrapib treatment in chow- and HFD-fed CETP-transgenic mice. -

An Abundant Dysfunctional Apolipoprotein A1 in Human Atheroma
Cleveland State University EngagedScholarship@CSU Mathematics Faculty Publications Mathematics Department 2-1-2014 An Abundant Dysfunctional Apolipoprotein A1 in Human Atheroma Ying Huang Cleveland Clinic Joseph A. DiDonato Cleveland State University, [email protected] Bruce S. Levison Cleveland Clinic Dave Schmitt Cleveland Clinic Lin Li Cleveland Clinic Follow this and additional works at: https://engagedscholarship.csuohio.edu/scimath_facpub Part of the Mathematics Commons See next page for additional authors How does access to this work benefit ou?y Let us know! Repository Citation Huang, Ying; DiDonato, Joseph A.; Levison, Bruce S.; Schmitt, Dave; Li, Lin; Wu, Yuping; Buffa, Jennifer; Kim, Timothy; Gerstenecker, Gary S.; Gu, Xiaodong; Kadiyala, Chandra S.; Wang, Zeneng; Culley, Miranda K.; Hazen, Jennie E.; DiDonato, Anthony J.; Fu, Xiaoming; Berisha, Stela Z.; Peng, Daoquan; Nguyen, Truc T.; Liang, Shaohong; Chuang, Chia-Chi; Cho, Leslie; PLow, Edward F.; Fox, Paul L.; Gogonea, Valentin; Tang, W.H. Wilson; Parks, John S.; Fisher, Edward A.; Smith, Jonathan D.; and Hazen, Stanley L., "An Abundant Dysfunctional Apolipoprotein A1 in Human Atheroma" (2014). Mathematics Faculty Publications. 161. https://engagedscholarship.csuohio.edu/scimath_facpub/161 This Article is brought to you for free and open access by the Mathematics Department at EngagedScholarship@CSU. It has been accepted for inclusion in Mathematics Faculty Publications by an authorized administrator of EngagedScholarship@CSU. For more information, please contact [email protected]. Authors Ying Huang, Joseph A. DiDonato, Bruce S. Levison, Dave Schmitt, Lin Li, Yuping Wu, Jennifer Buffa, Timothy Kim, Gary S. Gerstenecker, Xiaodong Gu, Chandra S. Kadiyala, Zeneng Wang, Miranda K. Culley, Jennie E. -

Coexpression of ATP-Binding Cassette Proteins ABCG5 and ABCG8 Permits Their Transport to the Apical Surface
Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface Gregory A. Graf, … , Jonathan C. Cohen, Helen H. Hobbs J Clin Invest. 2002;110(5):659-669. https://doi.org/10.1172/JCI16000. Article Cardiology Mutations in either ATP-binding cassette (ABC) G5 or ABCG8 cause sitosterolemia, an autosomal recessive disorder of sterol trafficking. To determine the site of action of ABCG5 and ABCG8, we expressed recombinant, epitope-tagged mouse ABCG5 and ABCG8 in cultured cells. Both ABCG5 and ABCG8 underwent N-linked glycosylation. When either protein was expressed individually in cells, the N-linked sugars remained sensitive to Endoglycosidase H (Endo H). When ABCG5 and ABCG8 were coexpressed, the attached sugars were Endo H–resistant and neuraminidase-sensitive, indicating that the proteins were transported to the trans-Golgi complex. The mature, glycosylated forms of ABCG5 and ABCG8 coimmunoprecipitated, consistent with heterodimerization of these two proteins. The Endo H–sensitive forms of ABCG5 and ABCG8 were confined to the endoplasmic reticulum (ER), whereas the mature forms were present in non-ER fractions in cultured hepatocytes. Immunoelectron microscopy revealed ABCG5 and ABCG8 on the plasma membrane of these cells. In polarized WIF-B cells, recombinant ABCG5 localized to the apical (canalicular) membrane when coexpressed with ABCG8, but not when expressed alone. To our knowledge this is the first direct demonstration that trafficking of an ABC half-transporter to the cell surface requires the presence of its dimerization partner. Find the latest version: https://jci.me/16000/pdf Coexpression of ATP-binding cassette See the related Commentary beginning on page 605. -

Cardiovascular Disease Products
Cardiovascular Disease Products For more information, visit: www.bosterbio.com Cardiovascular Disease Research Cardiovascular disease is the leading cause of death in developed nations. Boster Bio aims to supply researchers with high-quality antibodies and ELISA kits so they can make new discoveries and help save lives. In this catalogue you will find a comprehensive list of high-affinity Boster antibodies and high sensitivity Boster ELISA kits targeted at proteins associated with cardiovascular disease. Boster: The Fastest Growing About Bosterbio Antibody Company In 2015 Boster is an antibody manufacturer founded in 1993 by histologist Steven Xia. Over the past two decades, Boster and its products have been cited in over 20,000 publications and counting. The firm specializes in developing antibodies and ELISA kits that feature high affinity, Boster Bio received the CitaAb award for high specificity at affordable the greatest increase in number of prices. citations during 2015 than any other antibody manufacturer. Table of Contents Boster Cardiovascular Disease Related Antibodies…………..………..... 2 Boster Cardiovascular Disease Related ELISA Kits……………………..…. 9 1 High Affinity Boster Antibodies Boster supplies only the highest quality antibodies. Our high-affinity polyclonal and monoclonal antibodies are thoroughly validated by Western Blotting, Immunohistochemistry and ELISA. This is our comprehensive catalog of our antibody products related to cardiovascular disease, sorted in alphabetical order by target gene name. Catalog No Product Name -
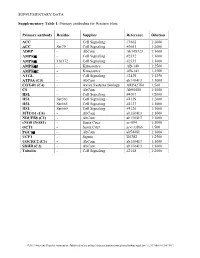
Supplementary Table 1
SUPPLEMENTARY DATA Supplementary Table 1. Primary antibodies for Western blots. Primary antibody Residue Supplier Reference Dilution ACC - Cell Signaling #3662 1:2000 ACC Ser79 Cell Signaling #3661 1:2000 ADRP - AbCam Ab108323 1:1000 AMPKα - Cell Signaling #2532 1:1000 AMPKα Thr172 Cell Signaling #2535 1:1000 AMPKα1 - Kinasource AB-140 1:2500 AMPKα2 - Kinasource AB-141 1:2500 ATGL - Cell Signaling #2439 1:1250 ATP5A (C5) - AbCam ab110413 1:1000 COX4l1 (C4) - Aviva Systems Biology ARP42784 1:500 CS - AbCam Ab96600 1:1000 HSL - Cell Signaling #4107 1:2000 HSL Ser563 Cell Signaling #4139 1:2000 HSL Ser565 Cell Signaling #4137 1:1000 HSL Ser660 Cell Signaling #4126 1:1000 MTCO1 (C4) - AbCam ab110413 1:1000 NDUFB8 (C1) - AbCam ab110413 1:1000 eNOS (NOS3) - Santa Cruz sc-654 1:1000 OCT1 - Santa Cruz sc-133866 1:500 PGC1α - AbCam ab54481 1:1000 UCP1 - Sigma U6382 1:2500 UQCRC2 (C3) - AbCam ab110413 1:1000 SDHB (C2) - AbCam ab110413 1:1000 Tubulin - Cell Signaling #2148 1:2000 ©2013 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0194/-/DC1 SUPPLEMENTARY DATA Supplementary Table 2. Primer sequences for qRT-PCR. Gene Accession number Forward primer Reverse primer Abca1 NM_013454.3 CCCAGAGCAAAAAGCGACTC GGTCATCATCACTTTGGTCCTTG Abcg5 NM_031884 TGTCCTACAGCGTCAGCAACC GGCCACTCTCGATGTACAAGG Abcg8 NM_026180 TCCTGTGAGCTGGGCATCCGA CCCGCAGCCTGAGCTCCCTAT Acaca NM_133360.2 CAGCTGGTGCAGAGGTACCG TCTACTCGCAGGTACTGCCG Acacb NM_133904.2 GCGCCTACTATGAGGCCCAGCA ACAAACTCGGCTGGGGACGC Acly NM_134037.2 -

Overexpression of ABCG5 and ABCG8 Promotes Biliary Cholesterol Secretion and Reduces Fractional Absorption of Dietary Cholestero
Overexpression of ABCG5 See the related Commentary beginning on page 605. and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol Liqing Yu,1 Jia Li-Hawkins,1 Robert E. Hammer,2,3 Knut E. Berge,1 Jay D. Horton,1 Jonathan C. Cohen,4 and Helen H. Hobbs1 1Department of Molecular Genetics and McDermott Center for Human Growth and Development, 2Department of Biochemistry, 3Howard Hughes Medical Institute, and 4Center for Human Nutrition, University of Texas Southwestern Medical Center at Dallas, Dallas, Texas, USA Two ATP-binding cassette (ABC) transporters, ABCG5 and ABCG8, have been proposed to limit sterol absorption and to promote biliary sterol excretion in humans. To test this hypothesis, a P1 clone containing the human ABCG5 and ABCG8 genes was used to generate transgenic mice. The transgenes were expressed primarily in the liver and small intestine, mirroring the expression pattern of the endogenous genes. Transgene expression only modestly affected plasma and liver cholesterol levels but profoundly altered cholesterol transport. The fractional absorption of dietary cholesterol was reduced by about 50%, and biliary cholesterol levels were increased more than fivefold. Fecal neu- tral sterol excretion was increased three- to sixfold and hepatic cholesterol synthesis increased two- to fourfold in the transgenic mice. No significant changes in the pool size, composition, and fecal excretion of bile acids were observed in the transgenic mice. Transgene expression attenuated the increase in hepatic cholesterol content induced by consumption of a high cholesterol diet. These results demonstrate that increased expression of ABCG5 and ABCG8 selectively drives biliary neutral sterol secretion and reduces intestinal cholesterol absorption, leading to a selective increase in neu- tral sterol excretion and a compensatory increase in cholesterol synthesis. -

Increased Phospholipid Transfer Protein Activity Associated with the Impaired Cellular Cholesterol Efflux in Type 2 Diabetic Subjects with Coronary Artery Disease
Tohoku J. Exp. Med., 2007,Cholesterol 213, 129-137 Efflux and PLTP Activity in Diabetes with CAD 129 Increased Phospholipid Transfer Protein Activity Associated with the Impaired Cellular Cholesterol Efflux in Type 2 Diabetic Subjects with Coronary Artery Disease 1,2 2 2 2 3 NEBIL ATTIA, AMEL NAKBI, MAHA SMAOUI, RAJA CHAABA, PHILIPPE MOULIN, 4 4 5 SONIA HAMMAMI, KHALDOUN BEN HAMDA, FRANÇOISE CHANUSSOT and 2 MOHAMED HAMMAMI 1Biology Department, Faculty of Sciences, University November 7th at Carthage, Bizerte, Tunisia 2Biochemistry Laboratory, UR 08-39, Faculty of Medicine, University of Monastir, Monastir, Tunisia 3U11 Cardiovascular Unit, Louis Pradel Hospital and INSERM U585, Lyon, France 4Departments of Internal Medicine (SH) and Cardiology (KBH), Monastir Hospital, Monastir, Tunisia 5INSERM U476, Faculty of Medecine la Timone, University Aix-Marseille II, Marseille, France ATTIA, N., NAKBI, A., SMAOUI, M., CHAABA, R., MOULIN, P., HAMMAMI, S., HAMDA, K.B., CHANUSSOT, F. and HAMMAMI, M. Increased Phospholipid Transfer Protein Activity Associated with the Impaired Cellular Cholesterol Efflux in Type 2 Diabetic Subjects with Coronary Artery Disease. Tohoku J. Exp. Med., 2007, 213 (2), 129-137 ── Reverse cholesterol transport (RCT) is the pathway, by which the excess of cholesterol is removed from peripheral cells to the liver. An early step of RCT is the efflux of free cholesterol from cell membranes that is mediated by high-density lipoproteins (HDL). Phospholipid transfer protein (PLTP) transfers phospholipids between apolipoprotein-B-containing lipo- proteins (i.e., chylomicrons and very low-density lipoproteins) and HDL. PLTP contrib- utes to the HDL maturation and increases the ability of HDL to extract the cellular choles- terol. -

A Case of Ezetimibe-Effective Hypercholesterolemia with a Novel Heterozygous Variant in ABCG5
2020, 67 (11), 1099-1105 Original A case of ezetimibe-effective hypercholesterolemia with a novel heterozygous variant in ABCG5 Yujiro Nakano1), Chikara Komiya1), Hitomi Shimizu2), 3), Hiroyuki Mishima3), Kumiko Shiba1), Kazutaka Tsujimoto1), Kenji Ikeda1), Kenichi Kashimada4), Sumito Dateki2), Koh-ichiro Yoshiura3), Yoshihiro Ogawa5) and Tetsuya Yamada1) 1) Department of Molecular Endocrinology and Metabolism, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo 113-8519, Japan 2) Department of Pediatrics, Graduate School of Biomedical Sciences, Nagasaki University, Nagasaski 852-8501, Japan 3) Department of Human Genetics, Graduate School of Biomedical Sciences, Nagasaki University, Nagasaki 852-8501, Japan 4) Department of Pediatrics and Developmental Biology, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo 113-8519, Japan 5) Department of Medicine and Bioregulatory Science, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan Abstract. Sitosterolemia is caused by homozygous or compound heterozygous gene mutations in either ATP-binding cassette subfamily G member 5 (ABCG5) or 8 (ABCG8). Since ABCG5 and ABCG8 play pivotal roles in the excretion of neutral sterols into feces and bile, patients with sitosterolemia present elevated levels of serum plant sterols and in some cases also hypercholesterolemia. A 48-year-old woman was referred to our hospital for hypercholesterolemia. She had been misdiagnosed with familial hypercholesterolemia at the age of 20 and her serum low-density lipoprotein cholesterol (LDL-C) levels had remained about 200–300 mg/dL at the former clinic. Although the treatment of hydroxymethylglutaryl-CoA (HMG-CoA) reductase inhibitors was ineffective, her serum LDL-C levels were normalized by ezetimibe, a cholesterol transporter inhibitor. -

Common Genetic Variations Involved in the Inter-Individual Variability Of
nutrients Review Common Genetic Variations Involved in the Inter-Individual Variability of Circulating Cholesterol Concentrations in Response to Diets: A Narrative Review of Recent Evidence Mohammad M. H. Abdullah 1 , Itzel Vazquez-Vidal 2, David J. Baer 3, James D. House 4 , Peter J. H. Jones 5 and Charles Desmarchelier 6,* 1 Department of Food Science and Nutrition, Kuwait University, Kuwait City 10002, Kuwait; [email protected] 2 Richardson Centre for Functional Foods & Nutraceuticals, University of Manitoba, Winnipeg, MB R3T 6C5, Canada; [email protected] 3 United States Department of Agriculture, Agricultural Research Service, Beltsville, MD 20705, USA; [email protected] 4 Department of Food and Human Nutritional Sciences, University of Manitoba, Winnipeg, MB R3T 2N2, Canada; [email protected] 5 Nutritional Fundamentals for Health, Vaudreuil-Dorion, QC J7V 5V5, Canada; [email protected] 6 Aix Marseille University, INRAE, INSERM, C2VN, 13005 Marseille, France * Correspondence: [email protected] Abstract: The number of nutrigenetic studies dedicated to the identification of single nucleotide Citation: Abdullah, M.M.H.; polymorphisms (SNPs) modulating blood lipid profiles in response to dietary interventions has Vazquez-Vidal, I.; Baer, D.J.; House, increased considerably over the last decade. However, the robustness of the evidence-based sci- J.D.; Jones, P.J.H.; Desmarchelier, C. ence supporting the area remains to be evaluated. The objective of this review was to present Common Genetic Variations Involved recent findings concerning the effects of interactions between SNPs in genes involved in cholesterol in the Inter-Individual Variability of metabolism and transport, and dietary intakes or interventions on circulating cholesterol concen- Circulating Cholesterol trations, which are causally involved in cardiovascular diseases and established biomarkers of Concentrations in Response to Diets: cardiovascular health.