Sorbitol As a Chain Extender of Polyurethane Prepolymers to Prepare Self-Healable and Robust Polyhydroxyurethane Elastomers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Study of the Partial and Exhaustive Intramolecular Dehydration of D-Sorbitol
Available online at http://journal-of-agroalimentary.ro Journal of Journal of Agroalimentary Processes and Agroalimentary Processes and Technologies 2013, 19(2), 259-270 Technologies Study of the partial and exhaustive intramolecular dehydration of D-sorbitol Ileana Cocan *, Monica Viorica Negrea, Ionel Vasile Jianu ”Dunărea de Jos” University of Galati, Faculty of Food Science and Engineering, Domnească Street, 47, RO-800008, Galati, Romania Received: 07 February 2013; Accepted: 09 March 2013 ______________________________________________________________________________________ Abstract Sweetener, humectants, sequestrant, texturizer, stabilizer, bulking agent, D-sorbitol represents a leading product in the processing of the polysorbate class. The scope of this work is to obtain details on the preparation and accession of D-sorbitol mono- and dianhydride as a method of chemical protection of primary and secondary hydroxyl functional groups (1:4) (3:6) as part of the strategy of controlled processing of polysorbates containing “homogeneous” polyoxyethylenic chains (n = 3, 6, 9, 18). This work is investigating the dependence of yield of internal dehydration (partial and/or exhaustive, direct of D-sorbitol) on temperature (100-160°C and 120-200°C , respectively ) and duration (5–35 minutes and 10–100 minutes , respectively ) for the processing of 1,4-sorbitan and 2,5-isosorbide. The evolution of the hydroxyl number (mg KOH/g D-sorbitol) (mg KOH/g 1,4-sorbitan) related to the theoretical (initial) value was followed and optimal values for the yield and dehydration parameters were established. Also in this work, the mathematical modeling of the dependence curves experimentally registered is realized. Keywords : izosorbide, D-sorbitol, D-glucitol, D-sorbit, 1,4-sorbitan ______________________________________________________________________________________ 1. -

Sorbitol Malabsorption 1/2
Internal Medicine and Gastroenterology Clinic Dres. Mares,M.Hanig,Mares, Hanig, Rambow, S.Blau, Blau, M.Seip, Hanig, Seip, A.Borchers,Blau,Kirchner, Hochstr. Hochstr. Hochstr. 43, 60313 43, 43, 60313 60313 Frankfurt/Main, Frankfurt/M., Frankfurt/M., www.gastroenterologie-ffm.de www.gastroenterologie-ffm.de www.gastroenterologie-ffm.de Nutrition hints for sorbitol malabsorption 1/2 Sorbitol intolerance Sorbitol incompatibility 1. What does this mean for nutrition? Avoid sorbitol as a sweetener and foods high in sorbitol. Small amount of sorbitol can often be tolerated (at most 10-10 g per day, sometimes less). In order to prevent general upper GI problems, easily digestible foods that do not cause gas are recommended. 2. Key points about foods – Avoid sorbitol and foods containing sorbitol – easily digestible food that does not cause gas 3. vegetables and fruits low in fibre (see 5) 4. Choice of foods The following foods are high in sorbitol and not suitable: – Sorbitol as sweetener: e.g. Sionon, Flarom, diabetic sweetener – Dietetic foods produced with sorbitol: for example, diabetic marmalades, diabetic sweets, diabetic baked goods – Types of fruit which have a naturally high sorbitol content: Apples, pears, cherries, prunes, plums, dates fruits with seeds, such as mirabelles, apricots, nectarines, and all fruit syrups made from these types of fruits – Types of fruit which have a naturally low sorbitol content: Berry fruits such as strawberries, raspberries, blackberries, blueberries, currants, gooseberries, citrus fruits, bananas, pineapples, kiwis – Sorbitol as a coating for: sultanas, raisins, and dried fruit or candied fruit – Sorbitol in sweets: chewing gum, jelly babies, jelly fruits, candies, chocolate bars, filled wafers, chocolate, etc. -

Copyrighted Material
Index Abbreviations receptor sites 202, 211 weak 122 amino acids, Table 500–1 muscarinic 413 weak, pH calculation 147–8 nucleic acids 551 nicotinic 413 Acid–base nucleotides, Table 551 as quaternary ammonium salt 202 catalysis, enzymes 516 peptides and proteins 504 Acetylcholinesterase equilibria 121 phosphates and diphosphates 277 enzyme mechanism 519–21 interactions, predicting 155–7 structural, Table 14 hydrolysis of acetylcholine 279 Acidic reagents, Table 157 ACE (angiotensin-converting enzyme) inhibitors 279 Acidity enzyme action 532 Acetyl-CoA carboxylase, in fatty acid acidity constant 122 inhibitors 532 biosynthesis 595 bond energy effects 125 Acetal Acetyl-CoA (acetyl coenzyme A) definition 121 in etoposide 233 as acylating agent 262 electronegativity effects 125 formation 229 carboxylation to malonyl-CoA 595, hybridization effects 128 polysaccharides as polyacetals 232 609 inductive effects 125–7 as protecting group 230 Claisen reaction 381 influence of electronic and structural Acetal and ketal enolate anions 373 features 125–34 cyclic, as protecting groups 481 in Krebs cycle 585 and leaving groups, Table 189 groups in sucrose 231 from β-oxidation of fatty acids 388 pKa values 122–5 Acetaldehyde, basicity 139 as thioester 262, 373 resonance / delocalization effects 129–34 Acetamide, basicity 139 Acetylene, bonding molecular orbitals 31 Acidity (compounds) Acetaminophen, see paracetamol Acetylenes, acidity 128 acetone 130 Acetoacetyl-CoA, biosynthesis from N-Acetylgalactosamine, in blood group acetonitrile 365 acetyl-CoA 392 -

Polyols Have a Variety of Functional Properties That Make Them Useful Alternatives to Sugars in Applications Including Baked Goods
Polyols have a variety of functional properties that make them useful alternatives to sugars in applications including baked goods. Photo © iStockphoto.com/Synergee pg 22 09.12 • www.ift.org BY LYN NABORS and THERESA HEDRICK SUGAR REDUCTION WITH Polyols Polyols are in a unique position to assist with reduced-sugar or sugar-free reformulations since they can reduce calories and complement sugar’s functionality. ugar reduction will be an important goal over the of the product’s original characteristics may still be main- next few years as consumers, government, and in- tained with the replacement of those sugars by polyols. Sdustry alike have expressed interest in lower-calorie In addition, excellent, good-tasting sugar-free products and lower-sugar foods. The 2010 Dietary Guidelines for can be developed by using polyols. Polyols are in a unique Americans put a strong emphasis on consuming fewer position to assist with reduced-sugar or sugar-free refor- calories and reducing intake of added sugars. The In- mulations; since they are only partially digested and ab- stitute of Medicine (IOM) held a public workshop in sorbed, they can reduce calories and complement sugar’s November 2010 to discuss ways the food industry can functionality. Polyols provide the same bulk as sugars and use contemporary and innovative food processing tech- other carbohydrates. Additionally, polyols have a clean, nologies to reduce calorie intake in an effort to reduce sweet taste, which is important since consumers are not and prevent obesity, and in October 2011 recommended likely to sacrifice taste for perceived health benefits. Poly- front-of-package labeling that includes rating the product ols have a host of other functional properties that make based on added sugars content. -

CHM 224 Test 2 Chapters 13, 14, Organometallics, 20 NAME
CHM 224 NAME: Test 2 Chapters 13, 14, organometallics, 20 1. Answer the following 3 questions: A. Brandy is 60% alcohol. What is its proof? B. This alcohol can be created by heating wood chips: C. This alcohol is referred to as "rubbing alcohol": 2. The three compounds below have nearly identical molecular weights. Arrange them according to their expected boiling points from highest >>> lowest. OH OH O A B C 3. Match the pKa values with the compounds provided: pKa's = 7.2, 8.0, 10.3 OH OH OH NO2 CH3 CN 4. What is the expected major product of the following reaction? 1. 1 equivalent BH3•THF 2. NaOH, H2O2 5. Which of the following compounds is expected to fail to react with KMnO4 (may be more than one)? A. 1-methylcyclopentanol B. 2-methyl-3-hexanol C. 4-ethyl-4-heptanol D. 3-bromo-1-butanol 6. Provide the IUPAC name for the following compound: OH Br 7. Which ONE of the following statements is true? A. ethers are generally water soluble, flammable, and reactive with strong bases B. ethers are generally water insoluble, not flammable, and reactive with strong acids C. ethers are generally water soluble, flammable, and reactive with strong bases D. ethers are generally water insoluble, flammable, and reactive with strong acids 8. Provide a synthetic route for the synthesis of tert-butyl isopropyl ether using the Williamson ether synthesis from an appropriate starting alcohol and alkyl halide. 9. Which one of the following alkenes will form the epoxide below upon treatment with a peroxyacid? O H CH2CH3 H3C CH3 A B C D 10. -

Pichia Process Optimization by Methanol/Sorbitol Co-Feeding – Patrick Fickers - University of Liege
Short Communication Journal of Applied Microbiology and Biochemistry 2020 Vol.4 No.4 Applied Microbiology 2016- Pichia process optimization by methanol/sorbitol co-feeding – Patrick Fickers - University of Liege Patrick Fickers University of Liege Abstract Recombinant protein production driven by AOX1 promoter is at this stage. During the second step, so as to extend biomass challenged by a high oxygen demand and warmth production, production and de-repress the methanol metabolic machinery, a especially in large-scale bioreactor. A reliable solution depends glycerol-limited fed-batch procedure is initiated. This phase on a methanol/sorbitol co-feeding strategy during the induction leads to gradual de-repression of the enzymes necessary for the phase. during this work, transient continuous cultures were first dissimilation of methanol and reduces the time necessary for performed to quantitatively assess the advantages of a the cells to adapt to growth on methanol. Finally, recombinant methanol/sorbitol co-feeding process with a P. pastoris Mut+ protein expression is typically induced by feeding methanol strain bearing a pAOX1-lacZ construct served as a reporter because the sole carbon source. gene. Our results demonstrated that cell-specific oxygen consumption (qO2) might be reduced by decreasing the A fermentation guideline for both Mut+ and Muts strains of P. methanol fraction within the feeding media. Optimal pAOX1 pastoris is available from Invitrogen . Stratton et al. devised a induction was achieved and maintained within the range of protocol for P. pastoris high cell-density fermentation, during 0.45~0.75 C-mol/C-mol of methanol fraction. additionally, the which detailed procedures are provided. -

Enhanced Protein Production by Sorbitol Co-Feeding with Methanol in Recombinant Pichia Pastoris Strains
Biotechnology and Bioprocess Engineering 22: 767-773 (2017) pISSN 1226-8372 DOI 10.1007/s12257-017-0011-9 eISSN 1976-3816 RESEARCH PAPER Enhanced Protein Production by Sorbitol Co-feeding with Methanol in Recombinant Pichia pastoris Strains Li Chen, Ali Mohsin, Ju Chu, Yingping Zhuang, Yamei Liu, and Meijin Guo Received: 9 January 2017 / Revised: 8 May 2017 / Accepted: 29 August 2017 © The Korean Society for Biotechnology and Bioengineering and Springer 2017 Abstract Pichia pastoris strains carrying 1, 6, 12, and 18 Keywords: sorbitol, co-feeding, multi-copy, Pichia pastoris, copies of the porcine insulin precursor (PIP) gene, were porcine insulin precursor employed to investigate the effects of sorbitol co-feeding with methanol on the physiology of the strains. Multicopy clones of the methylotrophic yeast were generated to vary 1. Introduction the PIP gene dosage and recombinant proteins. Elevated gene dosage increased levels of the recombinant PIP protein The methylotrophic yeast, Pichia pastoris, has been when methanol served as the sole carbon and energy developed to be a highly successful system for large-scale source i.e., an increase of 1.9% for a strain carrying 1 copy, production of functionally active recombinant proteins 42.6% for a strain carrying 6 copies, 34.7% for a strain [1,2]. As a yeast, it is a unique system which has many carrying 12 copies and 80.9% for a strain carrying 18 advantages e.g., its ability to produce foreign proteins at copies, respectively (using sorbitol co-feeding with methanol high levels using one of the strongest and highly regulated during the induction phase). -
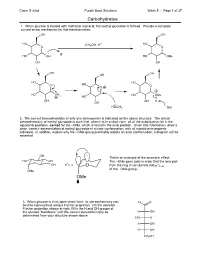
5.01 Carbohydrates Answers.Cdx
Chem S-20ab Purple Book Solutions Week 5 - Page 1 of 37 Carbohydrates 1. When glucose is treated with methanol and acid, the methyl glucoside is formed. Provide a complete curved-arrow mechanism for this transformation: OH OH HO CH OH, H+ HO O 3 O H+ HO OH HO OMe OH OH OH OH OH HO HO O HO O O HO OH HO OMe 2 HO OH OH H OH HOCH3 Sol 2. The correct stereochemistry of only one stereocenter is indicated on the above structure. The actual stereochemistry of methyl glucoside is such that, when it is in a chair form, all of the substituents fall in the equatorial positions, except for the –OMe, which is found in the axial position. Given this information, draw a clear, correct representation of methyl glucoside in a chair conformation, with all substituents properly indicated. In addition, explain why the –OMe group preferably adopts an axial conformation; a diagram will be essential. OH This is an example of the anomeric effect. HO OH The –OMe goes axial in order that the lone pair O * OH C–O O fromtheringOcandonateinto *C–O of the –OMe group. OMe OMe 3. When glucose is in its open-chain form, its stereochemistry can H O best be represented using a Fischer projection. On the skeleton Fischer projection shown at right, fill in the H and OH groups of the glucose "backbone" with the correct stereochemistry as H OH determined from your structure shown above. HO H H OH H OH CH2OH Chem S-20ab Purple Book Solutions Week 5 - Page 2 of 37 Carbohydrates: Mechanisms 1. -

Review on Artificial Sweeteners Used in Formulation of Sugar Free Syrups
International Journal of Advances in Pharmaceutics ISSN: 2320–4923; DOI: 10.7439/ijap Volume 4 Issue 2 [2015] Journal home page: http://ssjournals.com/index.php/ijap Review Article Review on artificial sweeteners used in formulation of sugar free syrups Afaque Raza Mehboob Ansari*, Saddamhusen Jahangir Mulla and Gosavi Jairam Pramod Department of Quality Assurance, D.S.T.S. Mandal’s College of Pharmacy, Jule Solapur-1, Bijapur Road, Solapur- 413004, Maharashtra, India. *Correspondence Info: Abstract Prof. Afaque Raza Mehboob Ansari Sweetening agents are employed in liquid formulations designed for oral Department of Quality Assurance, administration specifically to increase the palatability of the therapeutic agent. The D.S.T.S. Mandal’s College of main sweetening agents employed in oral preparations are sucrose, liquid glucose, Pharmacy, Jule Solapur-1, Bijapur glycerol, Sorbitol, saccharin sodium and aspartame. The use of artificial Road, Solapur- 413004, Maharashtra, sweetening agents in formulations is increasing and, in many formulations, India saccharin sodium is used either as the sole sweetening agent or in combination Email: [email protected] with sugars or Sorbitol to reduce the sugar concentration in the formulation. The Keywords: use of sugars in oral formulations for children and patients with diabetes mellitus is to be avoided. The present review discusses about the Artificial sweetening agents Sugar free syrup, which are generally used while the preparation of Sugar-free Syrup. Artificial sweeteners, Diabetes mellitus, Sucralose, and Aspartame. 1. Introduction Syrups are highly concentrated, aqueous solutions of sugar or a sugar substitute that traditionally contain a flavoring agent, e.g. cherry syrup, cocoa syrup, orange syrup, raspberry syrup. -

Hydrogenolysis of Sorbitol to Glycols Over Carbon Nanofibers-Supported Ruthenium Catalyst: the Role of Base Promoter
Chinese Journal of Catalysis 35 (2014) 692–702 available at www.sciencedirect.com journal homepage: www.elsevier.com/locate/chnjc Article (Special Issue on the 2nd International Congress on Catalysis for Biorefineries (CatBior 2013)) Hydrogenolysis of sorbitol to glycols over carbon nanofibers‐ supported ruthenium catalyst: The role of base promoter Jinghong Zhou *, Guocai Liu, Zhijun Sui, Xinggui Zhou, Weikang Yuan State Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China ARTICLE INFO ABSTRACT Article history: Sorbitol hydrogenolysis over carbon nanofibers‐supported Ru (Ru/CNFs) was carried out with Received 7 March 2014 different bases (NaOH, KOH, Mg(OH)2, Ba(OH)2, and CaO) to investigate the role of base promoter. Accepted 19 March 2014 The results indicated that all the bases used significantly enhanced the sorbitol conversion while the Published 20 May 2014 glycol selectivities varied with the base type and amount. CaO was the best base in terms of glycol selectivity for two reasons. CaO provided OH− for the base‐promoted cleavage of C–C bonds, while it 2+ Keywords: also supplied Ca for complexation with the intermediate aldehydes, thus affecting the reaction Sorbitol pathways. We identified an optimum ratio among sorbitol concentration, Ru/CNFs catalyst, and CaO Hydrogenolysis to achieve favorable glycol selectivities in sorbitol hydrogenolysis. Reaction pathways for sorbitol Base promoter hydrogenolysis into glycols in aqueous solution in the presence of CaO have been proposed based Glycol on the mechanistic study. Ruthenium catalyst © 2014, Dalian Institute of Chemical Physics, Chinese Academy of Sciences. Published by Elsevier B.V. All rights reserved. -

Gut Microbiota Prevents Sugar Alcohol-Induced Diarrhea
nutrients Article Gut Microbiota Prevents Sugar Alcohol-Induced Diarrhea Kouya Hattori 1,2 , Masahiro Akiyama 1, Natsumi Seki 1,2, Kyosuke Yakabe 1,2, Koji Hase 2 and Yun-Gi Kim 1,* 1 Research Center for Drug Discovery, Faculty of Pharmacy and Graduate School of Pharmaceutical Sciences, Keio University, Tokyo 105-8512, Japan; [email protected] (K.H.); [email protected] (M.A.); [email protected] (N.S.); [email protected] (K.Y.) 2 Department of Biochemistry, Faculty of Pharmacy and Graduate School of Pharmaceutical Sciences, Keio University, Tokyo 105-8512, Japan; [email protected] * Correspondence: [email protected] Abstract: While poorly-absorbed sugar alcohols such as sorbitol are widely used as sweeteners, they may induce diarrhea in some individuals. However, the factors which determine an individual’s susceptibility to sugar alcohol-induced diarrhea remain unknown. Here, we show that specific gut bacteria are involved in the suppression of sorbitol-induced diarrhea. Based on 16S rDNA analysis, the abundance of Enterobacteriaceae bacteria increased in response to sorbitol consumption. We found that Escherichia coli of the family Enterobacteriaceae degraded sorbitol and suppressed sorbitol-induced diarrhea. Finally, we showed that the metabolism of sorbitol by the E. coli sugar phosphotransferase system helped suppress sorbitol-induced diarrhea. Therefore, gut microbiota prevented sugar alcohol-induced diarrhea by degrading sorbitol in the gut. The identification of the gut bacteria which respond to and degrade sugar alcohols in the intestine has implications for microbiome science, processed food science, and public health. Keywords: gut microbiota; sugar alcohol; diarrhea Citation: Hattori, K.; Akiyama, M.; Seki, N.; Yakabe, K.; Hase, K.; Kim, Y.-G. -

Albendazole 40Mg/Ml Oral
SUGGESTED FORMULA ____________________________________________________________________________________ Albendazole 40mg/mL Oral Veterinary Suspension Version number: 1.0 Volume: 100mL ____________________________________________________________________________________ Albendazole, USP 4gm Simethicone, USP (S1926) 0.024gm Polysorbate 80, NF (PO138) 0.1gm Xanthan Gum, NF (XA105) 0.3gm Sucrose Crystals, NF (SU103) 39gm Sorbitol , NF (SO129) 13gm Sodium Benzoate, NF (SO120) 0.4gm Potassium Sorbate, NF (PO300) 0.4gm Citric Acid Monohydrate, USP (C1296) 0.06gm *Flavor XmL Purified Water, USP (W1014) Q.S. 100mL *Suggested flavors: Mixture of Vanilla and Orange Flavor SUGGESTED COMPOUNDING PROCEDURES 1. Calculate the required quantity of each ingredient for the total amount to be prepared 2. Accurately weigh and/or measure each ingredient 3. Heat 20mL purified water to 90° and add the sodium benzoate; allow to cool to 40°C 4. In a separate container, dissolve the citric acid in 15mL of purified water and combine the two aqueous solutions 5. In a separate container, heat another portion of purified water to 65°C and disperse the xanthan gum allowing to hydrate completely; allow to cool to room temperature 6. Combine the aqueous mixtures 7. Add the potassium sorbate and sucrose stirring until dissolved; mix well 8. Separately; mix the Albendazole with the Polysorbate 80 and sorbitol to a paste, add the suspension mixture and mix 9. Add the simethicone, flavor and sufficient purified water to volume; mix well 10. Package in glass and label 11. Suggested Quality Assessments – follow pharmacy SOPs: a. Weight to Volume calculation b. Color c. Pourability d. Settling e. Resuspendability Store in air tight amber glass container Store Room Temperature No claims are made as to the safety or efficacy of this preparation.