Enantioselective Assembly of Tertiary Stereocenters Via Multicomponent Chemoselective Cross- Cite This: Chem
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Stereochemistry, Alkyl Halide Substitution (SN1 & SN2)
Chem 261 Assignment & Lecture Outline 3: Stereochemistry, Alkyl Halide Substitution (SN1 & SN2) Read Organic Chemistry, Solomons, Fryle & Snyder 12th Edition (Electronic) • Functional Group List – Learn to recognize – Please see Green Handout – also p 76 of text • Periodic Table – Inside Front Cover - know 1st 10 elements (up through Neon) • Relative Strength of Acids and Bases – Inside Front cover (reference only) • Chapter 5 – Stereochemistry • Chapter 6 –Ionic Reactions: Nucleophilic Substitution of Alkyl Halides Problems: Do Not turn in, answers available in "Study Guide Student Solutions Manual " Solomons, Fryle, Snyder • Chapter 5: 5.1 to 5.15; 5.18 to 5.21; 5.26; 5.28; 5.33a-d; 5.46 • Chapter 6: 6.1 to 6.5; 6.7 to 6.10; 6.13; 6.20; 6.26; 6.27 Lecture Outline # 3 I. Comparison of 2 Structures: Same Molecular Formula ? -> If Yes, Possibly Isomers or Identical Same Arrangement (Sequence) of Groups ? If No -> Structural Isomers If Yes -> Superposable? If Yes -> Identical Structures If No -> Stereoisomers Non-Superposable Mirror Images ? If NO -> Diastereomers If Yes -> Enantiomers II. Chirality and Stereoisomers A. The Concept of Chirality 1. Identification of chiral objects a) achiral = not chiral b) planes of symmetry within a molecule 2. Types of stereoisomers – enantiomers and diastereomers B. Location of stereogenic (chiral) centres – 4 different groups on tetrahedral atom 1. Enantiomers & diastereomers 2. Meso compounds - chiral centers with plane of symmetry within molecule 3. Molecules with more than one chiral centre 4. Recognition of chiral centers in complex molecules - cholesterol - 8 chiral centres Drawing the enantiomer of cholesterol and its potential 255 stereoisomers 5. -

Supramolecular Chirality: Solvent Chirality Transfer in Molecular Chemistry and Polymer Chemistry
Symmetry 2014, 6, 677-703; doi:10.3390/sym6030677 OPEN ACCESS symmetry ISSN 2073-8994 www.mdpi.com/journal/symmetry Review Supramolecular Chirality: Solvent Chirality Transfer in Molecular Chemistry and Polymer Chemistry Michiya Fujiki Graduate School of Materials Science, Nara Institute of Science and Technology (NAIST), 8916-5 Takayama, Ikoma, Nara 630-0036, Japan; E-Mail: [email protected]; Tel.: +81-743-72-6040 Received: 15 July 2014; in revised form: 7 August 2014 / Accepted: 7 August 2014 / Published: 13 August 2014 Abstract: Controlled mirror symmetry breaking arising from chemical and physical origin is currently one of the hottest issues in the field of supramolecular chirality. The dynamic twisting abilities of solvent molecules are often ignored and unknown, although the targeted molecules and polymers in a fluid solution are surrounded by solvent molecules. We should pay more attention to the facts that mostly all of the chemical and physical properties of these molecules and polymers in the ground and photoexcited states are significantly influenced by the surrounding solvent molecules with much conformational freedom through non-covalent supramolecular interactions between these substances and solvent molecules. This review highlights a series of studies that include: (i) historical background, covering chiral NaClO3 crystallization in the presence of D-sugars in the late 19th century; (ii) early solvent chirality effects for optically inactive chromophores/fluorophores in the 1960s–1980s; and (iii) the recent development of mirror symmetry breaking from the corresponding achiral or optically inactive molecules and polymers with the help of molecular chirality as the solvent use quantity. Keywords: optically active; chiral; achiral; supramolecules; polymers; solvent; circular dichroism; circularly polarized luminescence; homochirality; molecular chirality 1. -

Alkyl Halides
Alkyl Halides Alkyl halides are a class of compounds where a halogen atom or atoms are bound to an sp3 orbital of an alkyl group. CHCl3 (Chloroform: organic solvent) CF2Cl2 (Freon-12: refrigerant CFC) CF3CHClBr (Halothane: anesthetic) Halogen atoms are more electronegative than carbon atoms, and so the C-Hal bond is polarized. The C-Hal (often written C-X) bond is polarized in such a way that there is partial positive charge on the carbon and partial negative charge on the halogen. Ch06 Alkyl Halides (landscape).docx Page 1 Dipole moment Electronegativities decrease in the order of: F > Cl > Br > I Carbon-halogen bond lengths increase in the order of: C-F < C-Cl < C-Br < C-I Bond Dipole Moments decrease in the order of: C-Cl > C-F > C-Br > C-I = 1.56D 1.51D 1.48D 1.29D Typically the chemistry of alkyl halides is dominated by this effect, and usually results in the C-X bond being broken (either in a substitution or elimination process). This reactivity makes alkyl halides useful chemical reagents. Ch06 Alkyl Halides (landscape).docx Page 2 Nomenclature According to IUPAC, alkyl halides are treated as alkanes with a halogen (Halo-) substituent. The halogen prefixes are Fluoro-, Chloro-, Bromo- and Iodo-. Examples: Often compounds of CH2X2 type are called methylene halides. (CH2Cl2 is methylene chloride). CHX3 type compounds are called haloforms. (CHI3 is iodoform). CX4 type compounds are called carbon tetrahalides. (CF4 is carbon tetrafluoride). Alkyl halides can be primary (1°), secondary (2°) or tertiary (3°). Other types: A geminal (gem) dihalide has two halogens on the same carbon. -

II. Nomenclature Rules for Alkenes 1. the Parent Name Will Be the Longest
1 Lecture 9 II. Nomenclature Rules For Alkenes 1. The parent name will be the longest carbon chain that contains both carbons of the double bond. Drop the -ane suffix of the alkane name and add the –ene suffix. Never name the double bond as a prefix. If a double bond is present, you have an alkene, not an alkane. alkane + -ene = alkene 2. Begin numbering the chain at the end nearest the double bond. Always number through the double bond and identify its position in the longest chain with the lower number. In the older IUPAC rules the number for the double bond was placed in front of the stem name with a hyphen. Under the newer rules, the number for the double bond is placed right in front of “ene”, with hyphens. We will use the newer rules for specifying the location of pi bonds. 1 2 3456 H3CCHCH CH 2 CH2 CH3 hex-2-ene (newer rules) 2-hexene (older rules) 3. Indicate the position of any substituent group by the number of the carbon atom in the parent (longest) chain to which it is attached. CH 1 2 345 3 H3CCHCHCHCH2 CH CH3 6 7 CH3 Numbering is determined by the double bond, not the branches, because the double bond has 5,6-dimethylhept-3-ene (newer rules) higher priority than any alkyl branch. 5,6-dimethyl-3-heptene (older rules) 4. Number cycloalkenes so that the double bond is 1,2 (number through the double bond). Number in the direction about the ring so that the lowest number is used at the first point of difference. -
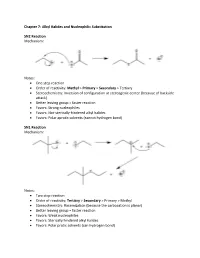
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Vicinal Dihalide: Two Halogen Atoms Are Bonded to Adjacent Carbons
DAMIETTA UNIVERSITY CHEM-103: BASIC ORGANIC CHEMISTRY LECTURE 4 Dr Ali El-Agamey Types of reactions 1- Addition reaction They normally involves unsaturated compounds capable of accepting additional atoms. 2- Substitution reaction Atom or group replaces atom or group. Types of reactions 3- Elimination reaction Atoms are removed to produce unsaturated compounds or ring. Organic Chemistry, 7th Edition L. G. Wade, Jr. Alkanes © 2010, Prentice Hall Hydrocarbons Hydrocarbons are molecules that are made of carbon and hydrogen ONLY. Alkanes • General formula: CnH2n+2 • Found in everything from natural gas to petroleum. • The smaller alkanes have very low boiling points (b.p.) therefore they are gases. • CH4 C2H6 C3H8 b.p. -160oC -89oC -42oC Preparation of Alkanes • 1- Hydrogenation of alkenes • 2- Alkyl halides (a) Via hydrolysis of Grignard reagent (b) Reduction by metal and acid (c) Coupling with organo-copper compounds. Preparation of Alkanes • (a) Hydrolysis of Grignard reagent Preparation of Alkanes • (b) Reduction by metal and acid Preparation of Alkanes • (c) Coupling with organo-copper compounds. i ' Preparation of Alkanes • (c) Coupling with organo-copper compounds. i Homework (1) Show how can you prepare n-butane from:1 (a) n-butyl bromide (b) sec-butyl bromide and (c) 2-butene. Complete the following equations:1 12 Chapter 8 Reactions of Alkanes • 1- Combustion • 2- Cracking (Pyrolysis) • 3- Halogenation – General reaction; examples – General mechanism; mechanism of specific example (CH4; propane) – Calculation of relative reactivities; -
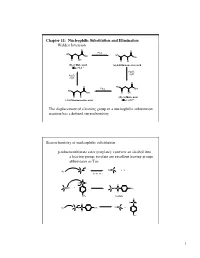
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion O O PCl5 HO HO OH OH O OH O Cl (S)-(-) Malic acid (+)-2-Chlorosuccinic acid [a]D= -2.3 ° Ag2O, H2O Ag2O, H2O O O HO PCl5 OH HO OH O OH O Cl (R)-(+) Malic acid (-)-2-Chlorosuccinic acid [a]D= +2.3 ° The displacement of a leaving group in a nucleophilic substitution reaction has a defined stereochemistry Stereochemistry of nucleophilic substitution p-toluenesulfonate ester (tosylate): converts an alcohol into a leaving group; tosylate are excellent leaving groups. abbreviates as Tos C X Nu C + X- Nu: X= Cl, Br, I O Cl S O O + C OH C O S CH3 O CH3 tosylate O -O S O O Nu C + Nu: C O S CH3 O CH3 1 O Tos-Cl - H3C O O H + TosO - H O H pyridine H O Tos O CH3 [a]D= +33.0 [a]D= +31.1 [a]D= -7.06 HO- HO- O - Tos-Cl H3C O - H O O TosO + H O H pyridine Tos H O CH3 [a]D= -7.0 [a]D= -31.0 [a]D= -33.2 The nucleophilic substitution reaction “inverts” the Stereochemistry of the carbon (electrophile)- Walden inversion Kinetics of nucleophilic substitution Reaction rate: how fast (or slow) reactants are converted into product (kinetics) Reaction rates are dependent upon the concentration of the reactants. (reactions rely on molecular collisions) H H Consider: HO C _ _ C Br Br HO H H H H At a given temperature: If [OH-] is doubled, then the reaction rate may be doubled If [CH3-Br] is doubled, then the reaction rate may be doubled A linear dependence of rate on the concentration of two reactants is called a second-order reaction (molecularity) 2 H H HO C _ _ C Br Br HO H H H H Reaction rates (kinetic) can be expressed mathematically: reaction rate = disappearance of reactants (or appearance of products) For the disappearance of reactants: - rate = k [CH3Br] [OH ] [CH3Br] = CH3Br concentration [OH-] = OH- concentration k= constant (rate constant) L mol•sec For the reaction above, product formation involves a collision between both reactants, thus the rate of the reaction is dependent upon the concentration of both. -

Chapter 21 Organic Chemistry
CHAPTER 21 ORGANIC CHEMISTRY Hydrocarbons 1. A hydrocarbon is a compound composed of only carbon and hydrogen. A saturated hydro- carbon has only carbon-carbon single bonds in the molecule. An unsaturated hydrocarbon has one or more carbon-carbon multiple bonds but may also contain carbon-carbon single bonds. A normal hydrocarbon has one chain of consecutively bonded carbon atoms. A branched hydrocarbon has at least one carbon atom not bonded to the end carbon of a chain of consecutively bonded carbon atoms. Instead, at least one carbon atom forms a bond to an inner carbon atom in the chain of consecutively bonded carbon atoms. 2. To determine the number of hydrogens bonded to the carbons in cyclic alkanes (or any alkane where they may have been omitted), just remember that each carbon has four bonds. In cycloalkanes, only the C−C bonds are shown. It is assumed you know that the remaining bonds on each carbon are C−H bonds. The number of C−H bonds is that number required to give the carbon four total bonds. 3. In order to form, cyclopropane and cyclobutane are forced to form bond angles much smaller than the preferred 109.5° bond angles. Cyclopropane and cyclobutane easily react in order to obtain the preferred 109.5° bond angles. 4. Aromatic hydrocarbons are a special class of unsaturated hydrocarbons based on the benzene ring. Benzene has the formula C6H6. It is a planar molecule (all atoms are in the same plane). Each carbon in benzene is attached to three other atoms; it exhibits trigonal planar geometry with 120° bond angles. -

Organic Chemistry (CHEM311) Fall 2005 Dr
Organic Chemistry (CHEM311) Fall 2005 Dr. Robert F. Dias ISOMERS: Same song, different dance. Isomer = “one of two or more compounds, radicals, or ions that contain the same number of atoms of the same elements but differ in structural arrangement and properties.” -Websters. You began learning about isomers in GenChem, now they are going to be integral to your understanding of organic chemistry. Below is a schematic showing the relationships between the different kinds of isomers that we are going to study… isomers structural stereoisomer (different bond pattern) (same bond pattern) enantiomer diastereomer conformers (non-superimposable (non-mirror images, (interconvertable on mirror image) non-superimposable) by bond rotation) geometric configurational (cis/trans) R,R vs. R,S. Various forms of this isomer tree appear in different books, but this is a good place to start. In this short section, we’ll look at the easy difference between structural isomers and all others. Consider pentane (n-, bp = 36.1 oC; mp = -129.8 oC). It has a molecular formula of C5H12…with 3 structural isomers: n-pentane, isopentane…neopentane 1 2 2 2 1 4 1 1 3 2 1 1 1 1 1 27 Organic Chemistry (CHEM311) Fall 2005 Dr. Robert F. Dias The numbers designate the primary (1o), secondary (2o), tertiary (3o), quaternary (4o) carbon…(dependent on how many other carbons are attached). There are 8 pentyl derivatives…3 n-pentyl, 4 isopentyl and 1 neopentyl, (if 1 substitution allowed) There are 5 structural isomers of hexane, 9 heptane isomers, but 18 octane isomers…yikes. C10 – 75 isomers; C15 – 4,347 isomers; C20 - 366,319 isomers…double yikes. -

Chem Soc Rev Tutorial Review
Please do not adjust margins Chem Soc Rev TUTORIAL REVIEW Stereoselective Arene Formation Achim Link and Christof Sparr* While aromatic hydrocarbons are ubiquitous in organic chemistry, they are typically not associated with chirality and stereoisomerism. Due to the planarity and symmetry of simple arenes, methods to assemble aromatic rings are therefore not routinely considered for the stereoselective synthesis of chiral compounds. The aim of this tutorial review is to contrast Received 15th December 2017, this common perception with the counterintuitive circumstance that stereoselective arene formation offers a means to Accepted stereoselectively prepare an exceptional range of chiral aromatic structures. The versatility of these methods across various types of molecular scaffolds allows to control stereocentre configuration, helical chiral compounds, the configuration of DOI: 10.1039/x0xx00000x rotationally restricted stereogenic axes, planar chiral molecules or curved polyaromatic systems. Furthermore, www.rsc.org/chemsocrev stereoselective arene formation holds great promise for the selective construction of extended- but structurally well-defined chiral structures. Key learning points: 1) Counterintuitively, arene formation offers a means to stereoselectively prepare a broad range of chiral structures. 2) Substrate desymmetrization by the de novo construction of an aromatic ring allows controlling stereocentre configuration 3) The non-planar shape of helicenes or curved aromatic systems can be controlled during arene assembly. 4) Rotationally restricted atropisomers and planar chiral compounds are readily accessible by stereoselective arene formation. forming reactions offer unique opportunities to prepare various 1. Introduction stereochemically complex compounds in isomerically enriched form, a concept that has recently gained momentum and Owing to the high symmetry of arene-archetypes such as emerged as an effective strategy also for the synthesis of benzene, naphthalene or anthracene, aromatic hydrocarbons extended structures. -

Infrared Tables (Short Summary of Common Absorption Frequencies)
Beauchamp Spectroscopy Tables 1 Infrared Tables (short summary of common absorption frequencies) The values given in the tables that follow are typical values. Specific bands may fall over a range of wavenumbers, cm-1. Specific substituents may cause variations in absorption frequencies. Absorption intensities may be stronger or weaker than expected, often depending on dipole moments. Additional bands may confuse the interpretation. In very symmetrical compounds there may be fewer than the expected number of absorption bands (it is even possible that all bands of a functional group may disappear, i.e. a symmetrically substituted alkyne!). Infrared spectra are generally informative about what functional groups are present, but not always. The 1H and 13C NMR’s are often just as informative about functional groups, and sometimes even more so in this regard. Information obtained from one spectroscopic technique should be verified or expanded by consulting the other spectroscopic techniques. IR Summary - All numerical values in the tables below are given in wavenumbers, cm-1 Bonds to Carbon (stretching wave numbers) sp3 C-X single bonds sp2 C-X single bonds CN CO CO CC CC CN 1000-1350 1050-1150 1100-1350 not used not very useful alkoxy C-O not very useful 1250 acyl and phenyl C-O sp2 C-X double bonds sp C-X triple bonds CO CC CN CC CN 1640-1810 expanded table 1600-1680 1640-1690 on next page 2100-2250 2240-2260 Stronger dipoles produce more intense IR bands and weaker dipoles produce less intense IR bands (sometimes none). Bonds to Hydrogen