Catenanes and Rotaxanes 10.7
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Stereochemistry, Alkyl Halide Substitution (SN1 & SN2)
Chem 261 Assignment & Lecture Outline 3: Stereochemistry, Alkyl Halide Substitution (SN1 & SN2) Read Organic Chemistry, Solomons, Fryle & Snyder 12th Edition (Electronic) • Functional Group List – Learn to recognize – Please see Green Handout – also p 76 of text • Periodic Table – Inside Front Cover - know 1st 10 elements (up through Neon) • Relative Strength of Acids and Bases – Inside Front cover (reference only) • Chapter 5 – Stereochemistry • Chapter 6 –Ionic Reactions: Nucleophilic Substitution of Alkyl Halides Problems: Do Not turn in, answers available in "Study Guide Student Solutions Manual " Solomons, Fryle, Snyder • Chapter 5: 5.1 to 5.15; 5.18 to 5.21; 5.26; 5.28; 5.33a-d; 5.46 • Chapter 6: 6.1 to 6.5; 6.7 to 6.10; 6.13; 6.20; 6.26; 6.27 Lecture Outline # 3 I. Comparison of 2 Structures: Same Molecular Formula ? -> If Yes, Possibly Isomers or Identical Same Arrangement (Sequence) of Groups ? If No -> Structural Isomers If Yes -> Superposable? If Yes -> Identical Structures If No -> Stereoisomers Non-Superposable Mirror Images ? If NO -> Diastereomers If Yes -> Enantiomers II. Chirality and Stereoisomers A. The Concept of Chirality 1. Identification of chiral objects a) achiral = not chiral b) planes of symmetry within a molecule 2. Types of stereoisomers – enantiomers and diastereomers B. Location of stereogenic (chiral) centres – 4 different groups on tetrahedral atom 1. Enantiomers & diastereomers 2. Meso compounds - chiral centers with plane of symmetry within molecule 3. Molecules with more than one chiral centre 4. Recognition of chiral centers in complex molecules - cholesterol - 8 chiral centres Drawing the enantiomer of cholesterol and its potential 255 stereoisomers 5. -

Weak Functional Group Interactions Revealed Through Metal-Free Active Template Rotaxane Synthesis
ARTICLE https://doi.org/10.1038/s41467-020-14576-7 OPEN Weak functional group interactions revealed through metal-free active template rotaxane synthesis Chong Tian 1,2, Stephen D.P. Fielden 1,2, George F.S. Whitehead 1, Iñigo J. Vitorica-Yrezabal1 & David A. Leigh 1* 1234567890():,; Modest functional group interactions can play important roles in molecular recognition, catalysis and self-assembly. However, weakly associated binding motifs are often difficult to characterize. Here, we report on the metal-free active template synthesis of [2]rotaxanes in one step, up to 95% yield and >100:1 rotaxane:axle selectivity, from primary amines, crown ethers and a range of C=O, C=S, S(=O)2 and P=O electrophiles. In addition to being a simple and effective route to a broad range of rotaxanes, the strategy enables 1:1 interactions of crown ethers with various functional groups to be characterized in solution and the solid state, several of which are too weak — or are disfavored compared to other binding modes — to be observed in typical host–guest complexes. The approach may be broadly applicable to the kinetic stabilization and characterization of other weak functional group interactions. 1 Department of Chemistry, University of Manchester, Manchester M13 9PL, UK. 2These authors contributed equally: Chong Tian, Stephen D. P. Fielden. *email: [email protected] NATURE COMMUNICATIONS | (2020) 11:744 | https://doi.org/10.1038/s41467-020-14576-7 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-14576-7 he bulky axle end-groups of rotaxanes mechanically lock To explore the scope of this unexpected method of rotaxane Trings onto threads, preventing the dissociation of the synthesis, here we carry out a study of the reaction with a series of components even if the interactions between them are not related electrophiles. -

The Emergence of Mass Spectrometry for Characterizing Nanomaterials
The emergence of mass spectrometry for characterizing nanomaterials. Atomically precise nanoclusters and beyond Clothilde Zerbino, Xavier Dagany, Fabien Chirot, Philippe Dugourd, Rodolphe Antoine To cite this version: Clothilde Zerbino, Xavier Dagany, Fabien Chirot, Philippe Dugourd, Rodolphe Antoine. The emergence of mass spectrometry for characterizing nanomaterials. Atomically precise nanoclus- ters and beyond. Materials Advances, Royal Society of Chemistry, 2021, 2 (15), pp.4896-4913. 10.1039/D1MA00261A. hal-03234241 HAL Id: hal-03234241 https://hal.archives-ouvertes.fr/hal-03234241 Submitted on 8 Jun 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Materials Advances View Article Online PERSPECTIVE View Journal The emergence of mass spectrometry for characterizing nanomaterials. Atomically precise Cite this: DOI: 10.1039/d1ma00261a nanoclusters and beyond Clothilde Comby-Zerbino, Xavier Dagany, Fabien Chirot, Philippe Dugourd and Rodolphe Antoine * Mass spectrometry (MS) is widely used in molecular science, and is now emerging as a characterization technique for ultra-small nanoparticles. In the field of atomically precise nanoclusters, MS combined to ionization sources such as electrospray and matrix-assisted laser desorption-ionization allows for accurate characterization of the size and charge state of the metal core and of the number of ligands. -

Supramolecular Chirality: Solvent Chirality Transfer in Molecular Chemistry and Polymer Chemistry
Symmetry 2014, 6, 677-703; doi:10.3390/sym6030677 OPEN ACCESS symmetry ISSN 2073-8994 www.mdpi.com/journal/symmetry Review Supramolecular Chirality: Solvent Chirality Transfer in Molecular Chemistry and Polymer Chemistry Michiya Fujiki Graduate School of Materials Science, Nara Institute of Science and Technology (NAIST), 8916-5 Takayama, Ikoma, Nara 630-0036, Japan; E-Mail: [email protected]; Tel.: +81-743-72-6040 Received: 15 July 2014; in revised form: 7 August 2014 / Accepted: 7 August 2014 / Published: 13 August 2014 Abstract: Controlled mirror symmetry breaking arising from chemical and physical origin is currently one of the hottest issues in the field of supramolecular chirality. The dynamic twisting abilities of solvent molecules are often ignored and unknown, although the targeted molecules and polymers in a fluid solution are surrounded by solvent molecules. We should pay more attention to the facts that mostly all of the chemical and physical properties of these molecules and polymers in the ground and photoexcited states are significantly influenced by the surrounding solvent molecules with much conformational freedom through non-covalent supramolecular interactions between these substances and solvent molecules. This review highlights a series of studies that include: (i) historical background, covering chiral NaClO3 crystallization in the presence of D-sugars in the late 19th century; (ii) early solvent chirality effects for optically inactive chromophores/fluorophores in the 1960s–1980s; and (iii) the recent development of mirror symmetry breaking from the corresponding achiral or optically inactive molecules and polymers with the help of molecular chirality as the solvent use quantity. Keywords: optically active; chiral; achiral; supramolecules; polymers; solvent; circular dichroism; circularly polarized luminescence; homochirality; molecular chirality 1. -

Alkyl Halides
Alkyl Halides Alkyl halides are a class of compounds where a halogen atom or atoms are bound to an sp3 orbital of an alkyl group. CHCl3 (Chloroform: organic solvent) CF2Cl2 (Freon-12: refrigerant CFC) CF3CHClBr (Halothane: anesthetic) Halogen atoms are more electronegative than carbon atoms, and so the C-Hal bond is polarized. The C-Hal (often written C-X) bond is polarized in such a way that there is partial positive charge on the carbon and partial negative charge on the halogen. Ch06 Alkyl Halides (landscape).docx Page 1 Dipole moment Electronegativities decrease in the order of: F > Cl > Br > I Carbon-halogen bond lengths increase in the order of: C-F < C-Cl < C-Br < C-I Bond Dipole Moments decrease in the order of: C-Cl > C-F > C-Br > C-I = 1.56D 1.51D 1.48D 1.29D Typically the chemistry of alkyl halides is dominated by this effect, and usually results in the C-X bond being broken (either in a substitution or elimination process). This reactivity makes alkyl halides useful chemical reagents. Ch06 Alkyl Halides (landscape).docx Page 2 Nomenclature According to IUPAC, alkyl halides are treated as alkanes with a halogen (Halo-) substituent. The halogen prefixes are Fluoro-, Chloro-, Bromo- and Iodo-. Examples: Often compounds of CH2X2 type are called methylene halides. (CH2Cl2 is methylene chloride). CHX3 type compounds are called haloforms. (CHI3 is iodoform). CX4 type compounds are called carbon tetrahalides. (CF4 is carbon tetrafluoride). Alkyl halides can be primary (1°), secondary (2°) or tertiary (3°). Other types: A geminal (gem) dihalide has two halogens on the same carbon. -

II. Nomenclature Rules for Alkenes 1. the Parent Name Will Be the Longest
1 Lecture 9 II. Nomenclature Rules For Alkenes 1. The parent name will be the longest carbon chain that contains both carbons of the double bond. Drop the -ane suffix of the alkane name and add the –ene suffix. Never name the double bond as a prefix. If a double bond is present, you have an alkene, not an alkane. alkane + -ene = alkene 2. Begin numbering the chain at the end nearest the double bond. Always number through the double bond and identify its position in the longest chain with the lower number. In the older IUPAC rules the number for the double bond was placed in front of the stem name with a hyphen. Under the newer rules, the number for the double bond is placed right in front of “ene”, with hyphens. We will use the newer rules for specifying the location of pi bonds. 1 2 3456 H3CCHCH CH 2 CH2 CH3 hex-2-ene (newer rules) 2-hexene (older rules) 3. Indicate the position of any substituent group by the number of the carbon atom in the parent (longest) chain to which it is attached. CH 1 2 345 3 H3CCHCHCHCH2 CH CH3 6 7 CH3 Numbering is determined by the double bond, not the branches, because the double bond has 5,6-dimethylhept-3-ene (newer rules) higher priority than any alkyl branch. 5,6-dimethyl-3-heptene (older rules) 4. Number cycloalkenes so that the double bond is 1,2 (number through the double bond). Number in the direction about the ring so that the lowest number is used at the first point of difference. -
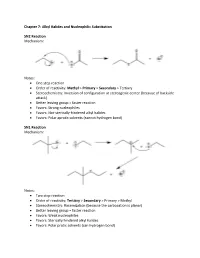
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Enantioselective Assembly of Tertiary Stereocenters Via Multicomponent Chemoselective Cross- Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Enantioselective assembly of tertiary stereocenters via multicomponent chemoselective cross- Cite this: Chem. Sci.,2016,7,2762 coupling of geminal chloro(iodo)alkanes† X. Jiang, K. Kulbitski, G. Nisnevich and M. Gandelman* Tertiary stereocenters represent a ubiquitous and highly important motif in many pharmaceutically active compounds and natural products. Development of efficient and straightforward approaches to their enantioselective construction from readily available simple substrates is an important yet challenging goal for synthetic chemistry. Herein we describe an efficient, versatile and facile method for the highly enantioselective construction of tertiary stereocenters via unprecedented consecutive one-pot Suzuki reactions of non-activated racemic 1-chloro-1-iodoalkanes with alkylboranes. It represents the first cross-coupling approach which employs simple and readily available primary alkyl substrates for the Received 16th November 2015 direct multicomponent assembly of enantioenriched tertiary stereocenters. A simple and effective Accepted 6th January 2016 Creative Commons Attribution 3.0 Unported Licence. preparation of 1-chloro-1-iodoalkanes from ubiquitous a-chloroalkanoic acids is also described. DOI: 10.1039/c5sc04378f Collectively, the developed methods open a door to efficient catalytic enantioselective synthesis of www.rsc.org/chemicalscience alkanes bearing tertiary stereocenters from carboxylic acids just in few steps. Introduction (electrophile -
Sir James Fraser Stoddart Baran Lab GM 2010-08-14
Y. Ishihara Sir James Fraser Stoddart Baran Lab GM 2010-08-14 (The UCLA USJ, 2007, 20, 1–7.) 1 Y. Ishihara Sir James Fraser Stoddart Baran Lab GM 2010-08-14 (The UCLA USJ, 2007, 20, 1–7.) 2 Y. Ishihara Sir James Fraser Stoddart Baran Lab GM 2010-08-14 Professor Stoddart's publication list (also see his website for a 46-page publication list): - 9 textbooks and monographs - 13 patents "Chemistry is for people - 894 communications, papers and reviews (excluding book chapters, conference who like playing with Lego abstracts and work done before his independent career, the tally is about 770) and solving 3D puzzles […] - At age 68, he is still very active – 22 papers published in the year 2010, 8 months in! Work is just like playing - He has many publications in so many fields... with toys." - Journals with 10+ papers: JACS 75 Acta Crystallogr Sect C 26 ACIEE 67 JCSPT1 23 "There is a lot of room for ChemEurJ 62 EurJOC 19 creativity to be expressed JCSCC 51 ChemComm 15 in chemis try by someone TetLett 42 Carbohydr Res 12 who is bent on wanting to OrgLett 35 Pure and Appl Chem 11 be inventive and make JOC 28 discoveries." - High-profile general science journals: Nature 4 Science 5 PNAS 8 - Reviews: AccChemRes 8 ChemRev 4 ChemSocRev 6 - Uncommon venues of publication for British or American scientists: Coll. Czechoslovak Chem. Comm. 5 Mendeleev Communications 2 Israel Journal of Chemistry 5 Recueil des Trav. Chim. des Pays-Bas 2 Canadian Journal of Chemistry 4 Actualité chimique 1 Bibliography (also see his website, http://stoddart.northwestern.edu/ , for a 56-page CV): Chemistry – An Asian Journal 3 Bulletin of the Chem. -

Rotaxanes and Catenanes by Click Chemistry
Mini Review Rotaxanes and Catenanes by Click Chemistry Ognjen Sˇ. Miljanic´a, William R. Dichtela, b, Ivan Aprahamiana, Rosemary D. Rohdeb, Heather D. Agnewb, James R. Heathb* and J. Fraser Stoddarta* a California NanoSystems Institute and Department of Chemistry and Biochemistry, University of California, Los Angeles, 405 Hilgard Avenue, Los Angeles, California 90095, USA, E-mail: [email protected] b Division of Chemistry and Chemical Engineering, California Institute of Technology, 1200 East California Boulevard, Pasadena, CA 91125, USA, E-mail: [email protected] Keywords: Catenanes, Click chemistry, Interlocked molecules, Rotaxanes, Self-assembly, Surface chemistry Received: June 1, 2007; Accepted: July 11, 2007 DOI: 10.1002/qsar.200740070 Abstract Copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition between terminal alkynes and azides – also known as the copper (Cu)-catalyzed Azide-Alkyne Cycloaddition (CuAAC) – has been used in the syntheses of molecular compounds with diverse structures and functions, owing to its functional group tolerance, facile execution, and mild reaction conditions under which it can be promoted. Recently, rotaxanes of four different structural types, as well as donor/acceptor catenanes, have been prepared using CuAAC, attesting to its tolerance to supramolecular interactions as well. In one instance of a rotaxane synthesis, the catalytic role of copper has been combined successfully with its previously documented ability to preorganize rotaxane precursors, i.e., form pseudoro- taxanes. The crystal structure of a donor/acceptor catenane formed using the CuAAC reaction indicates that any secondary [p···p] interactions between the 1,2,3-triazole ring and the bipyridinium p-acceptor are certainly not destabilizing. Finally, the preparation of robust rotaxane and catenane molecular monolayers onto metal and semiconductor surfaces is premeditated based upon recent advances in the use of the Huisgen reaction for surface functionalization. -

How Molecules Became Machines
THE NOBEL PRIZE IN CHEMISTRY 2016 POPULAR SCIENCE BACKGROUND How molecules became machines The Nobel Prize in Chemistry 2016 is awarded to Jean-Pierre Sauvage, Sir J. Fraser Stoddart and Bernard L. Feringa for their development of molecular machines that are a thousand times thinner than a hair strand. This is the story of how they succeeded in linking molecules together to design everything from a tiny lift to motors and miniscule muscles. How small can you make machinery? This is the question that Nobel Laureate Richard Feynman, famed for his 1950s’ predictions of developments in nanotechnology, posed at the start of a visionary lecture in 1984. Barefoot, and wearing a pink polo top and beige shorts, he turned to the audience and said: “Now let us talk about the possibility of making machines with movable parts, which are very tiny.” He was convinced it was possible to build machines with dimensions on the nanometre scale. These already existed in nature. He gave bacterial flagella as an example, corkscrew-shaped macromole- cules which, when they spin, make bacteria move forward. But could humans – with their gigantic hands – build machines so small that you would need an electron microscope to see them? A vision of the future – molecular machines will exist within 25–30 years One possible way would be to build a pair of mechanical hands that are smaller than your own, which in turn build a pair of smaller hands, which build even smaller hands, and so on, until a pair of miniscule hands can build equally miniscule machinery.