Supramolecular Chirality: Solvent Chirality Transfer in Molecular Chemistry and Polymer Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sensors 2010, 10, 6796-6820; Doi:10.3390/S100706796 OPEN ACCESS Sensors ISSN 1424-8220
Sensors 2010, 10, 6796-6820; doi:10.3390/s100706796 OPEN ACCESS sensors ISSN 1424-8220 www.mdpi.com/journal/sensors Review Intelligent Chiral Sensing Based on Supramolecular and Interfacial Concepts Katsuhiko Ariga 1,2,*, Gary J. Richards 1, Shinsuke Ishihara 1, Hironori Izawa 1,2 and Jonathan P. Hill 1,2 1 World Premier International (WPI) Research Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba 305-0044, Japan 2 Japan Science and Technology Agency, CREST, 1-1 Namiki, Tsukuba 305-0044, Japan * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +81-29-860-4597; Fax: +81-29-860-4832. Received: 17 June 2010; in revised form: 7 July 2010 / Accepted: 8 July 2010 / Published: 13 July 2010 Abstract: Of the known intelligently-operating systems, the majority can undoubtedly be classed as being of biological origin. One of the notable differences between biological and artificial systems is the important fact that biological materials consist mostly of chiral molecules. While most biochemical processes routinely discriminate chiral molecules, differentiation between chiral molecules in artificial systems is currently one of the challenging subjects in the field of molecular recognition. Therefore, one of the important challenges for intelligent man-made sensors is to prepare a sensing system that can discriminate chiral molecules. Because intermolecular interactions and detection at surfaces are respectively parts of supramolecular chemistry -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Stereochemistry, Alkyl Halide Substitution (SN1 & SN2)
Chem 261 Assignment & Lecture Outline 3: Stereochemistry, Alkyl Halide Substitution (SN1 & SN2) Read Organic Chemistry, Solomons, Fryle & Snyder 12th Edition (Electronic) • Functional Group List – Learn to recognize – Please see Green Handout – also p 76 of text • Periodic Table – Inside Front Cover - know 1st 10 elements (up through Neon) • Relative Strength of Acids and Bases – Inside Front cover (reference only) • Chapter 5 – Stereochemistry • Chapter 6 –Ionic Reactions: Nucleophilic Substitution of Alkyl Halides Problems: Do Not turn in, answers available in "Study Guide Student Solutions Manual " Solomons, Fryle, Snyder • Chapter 5: 5.1 to 5.15; 5.18 to 5.21; 5.26; 5.28; 5.33a-d; 5.46 • Chapter 6: 6.1 to 6.5; 6.7 to 6.10; 6.13; 6.20; 6.26; 6.27 Lecture Outline # 3 I. Comparison of 2 Structures: Same Molecular Formula ? -> If Yes, Possibly Isomers or Identical Same Arrangement (Sequence) of Groups ? If No -> Structural Isomers If Yes -> Superposable? If Yes -> Identical Structures If No -> Stereoisomers Non-Superposable Mirror Images ? If NO -> Diastereomers If Yes -> Enantiomers II. Chirality and Stereoisomers A. The Concept of Chirality 1. Identification of chiral objects a) achiral = not chiral b) planes of symmetry within a molecule 2. Types of stereoisomers – enantiomers and diastereomers B. Location of stereogenic (chiral) centres – 4 different groups on tetrahedral atom 1. Enantiomers & diastereomers 2. Meso compounds - chiral centers with plane of symmetry within molecule 3. Molecules with more than one chiral centre 4. Recognition of chiral centers in complex molecules - cholesterol - 8 chiral centres Drawing the enantiomer of cholesterol and its potential 255 stereoisomers 5. -

Chiral Metal–Macrocycle Frameworks: Supramolecular Chirality Induction and Helicity Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Chiral metal–macrocycle frameworks: supramolecular chirality induction and helicity Cite this: Chem. Sci.,2016,7, 2217 inversion of the helical macrocyclic structures† Ryou Kubota, Shohei Tashiro and Mitsuhiko Shionoya* Porous molecular solids composed of discrete macrocycles/cages have great potential for catalysis, separation and sensing techniques. Dynamic structural transformation of the host building blocks, especially a helicity inversion responsive to chemical triggers, is central to upgrading the spatial functions. Here we have achieved the syntheses of homochiral porous molecular solids composed of helical metal macrocycles through supramolecular chirality induction to both enantiomorphic forms with Received 27th November 2015 the aid of two different enantiopure sugar-derived lactones in the crystallization process. Moreover, we Accepted 23rd December 2015 found that the helicity of the macrocyclic skeletons can be inverted in the crystalline state only by DOI: 10.1039/c5sc04570c changing the type of solvent. This finding would lead to dynamic control of space chirality in connection Creative Commons Attribution 3.0 Unported Licence. www.rsc.org/chemicalscience with optical resolution, chiral amplification and asymmetric reactions. Introduction ions normally affords racemic crystals containing an equal amount of (M)- and (P)-forms of helical metal–macrocycles. Porous molecular solids (PMSs) composed of shape-persistent Moreover, it is difficult to regulate asymmetric crystallization macrocycles/cages show promise for solid-state functions from an equilibrated mixture of helical macrocycles in such as molecular storage, separation and catalysis.1,2 a predictable manner, and it is even more challenging to Through weak intermolecular interactionssuchasvander control the chirality of porous crystals in the solid state. -

Alkyl Halides
Alkyl Halides Alkyl halides are a class of compounds where a halogen atom or atoms are bound to an sp3 orbital of an alkyl group. CHCl3 (Chloroform: organic solvent) CF2Cl2 (Freon-12: refrigerant CFC) CF3CHClBr (Halothane: anesthetic) Halogen atoms are more electronegative than carbon atoms, and so the C-Hal bond is polarized. The C-Hal (often written C-X) bond is polarized in such a way that there is partial positive charge on the carbon and partial negative charge on the halogen. Ch06 Alkyl Halides (landscape).docx Page 1 Dipole moment Electronegativities decrease in the order of: F > Cl > Br > I Carbon-halogen bond lengths increase in the order of: C-F < C-Cl < C-Br < C-I Bond Dipole Moments decrease in the order of: C-Cl > C-F > C-Br > C-I = 1.56D 1.51D 1.48D 1.29D Typically the chemistry of alkyl halides is dominated by this effect, and usually results in the C-X bond being broken (either in a substitution or elimination process). This reactivity makes alkyl halides useful chemical reagents. Ch06 Alkyl Halides (landscape).docx Page 2 Nomenclature According to IUPAC, alkyl halides are treated as alkanes with a halogen (Halo-) substituent. The halogen prefixes are Fluoro-, Chloro-, Bromo- and Iodo-. Examples: Often compounds of CH2X2 type are called methylene halides. (CH2Cl2 is methylene chloride). CHX3 type compounds are called haloforms. (CHI3 is iodoform). CX4 type compounds are called carbon tetrahalides. (CF4 is carbon tetrafluoride). Alkyl halides can be primary (1°), secondary (2°) or tertiary (3°). Other types: A geminal (gem) dihalide has two halogens on the same carbon. -

Chiral Solvation Induced Supramolecular Chiral Assembly of Achiral Polymers
Chapter 4 Chiral Solvation Induced Supramolecular Chiral Assembly of Achiral Polymers Wei Zhang, Yin Zhao and Lu Yin Additional information is available at the end of the chapter http://dx.doi.org/10.5772/67700 Abstract To date, liquid crystal chirality, mechanophysical chirality, circularly polarized photon chirality, gelation and chiral solvation are all feasible candidates to generate optically active polymers and supramolecular chirality when employing achiral molecules as start‐ ing substances. Among this, chiral‐solvation‐induced chirality is one of the dominant methods for construction of chirality from achiral sources, such as achiral poly(n‐hexyl isocyanate) (PHIC), π‐conjugated polymers, oligo(p‐phenylenevinylene), polyacetylenes, σ‐conjugated polysilanes and side‐chain polymers. Supramolecular chirality is well established through their intra‐ or inter‐molecular noncovalent interactions, such as van der Waals, CH/π, dipole‐dipole interactions, hydrogen bonding and metal‐ligand coordi‐ nating interactions. Compared with the traditional methods, this strategy avoids the use of expensive chiral reagents and also expands the scope towards challenging substrates. This chapter highlights a series of studies that include: (i) the development‐historical background of chiral solvent induction strategy; (ii) the chiral‐solvation‐induced chirality in small molecules and oligomers; and (iii) recent developments in polymers, especially in π‐conjugated polymers and σ‐conjugated polymers. Keywords: optical activity, supramolecular chirality, chiral solvation, self‐assembly, circular dichroism, circularly polarized luminescence 1. Introduction As early as the second half of the nineteenth century, many scientists have long thought that the intrinsic biomolecular homochirality found in the living world is the origin of life on earth, since inherent optical activity exists inside all living organisms [1–14]. -

II. Nomenclature Rules for Alkenes 1. the Parent Name Will Be the Longest
1 Lecture 9 II. Nomenclature Rules For Alkenes 1. The parent name will be the longest carbon chain that contains both carbons of the double bond. Drop the -ane suffix of the alkane name and add the –ene suffix. Never name the double bond as a prefix. If a double bond is present, you have an alkene, not an alkane. alkane + -ene = alkene 2. Begin numbering the chain at the end nearest the double bond. Always number through the double bond and identify its position in the longest chain with the lower number. In the older IUPAC rules the number for the double bond was placed in front of the stem name with a hyphen. Under the newer rules, the number for the double bond is placed right in front of “ene”, with hyphens. We will use the newer rules for specifying the location of pi bonds. 1 2 3456 H3CCHCH CH 2 CH2 CH3 hex-2-ene (newer rules) 2-hexene (older rules) 3. Indicate the position of any substituent group by the number of the carbon atom in the parent (longest) chain to which it is attached. CH 1 2 345 3 H3CCHCHCHCH2 CH CH3 6 7 CH3 Numbering is determined by the double bond, not the branches, because the double bond has 5,6-dimethylhept-3-ene (newer rules) higher priority than any alkyl branch. 5,6-dimethyl-3-heptene (older rules) 4. Number cycloalkenes so that the double bond is 1,2 (number through the double bond). Number in the direction about the ring so that the lowest number is used at the first point of difference. -
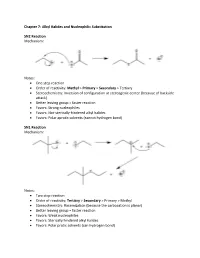
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Enantioselective Assembly of Tertiary Stereocenters Via Multicomponent Chemoselective Cross- Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Enantioselective assembly of tertiary stereocenters via multicomponent chemoselective cross- Cite this: Chem. Sci.,2016,7,2762 coupling of geminal chloro(iodo)alkanes† X. Jiang, K. Kulbitski, G. Nisnevich and M. Gandelman* Tertiary stereocenters represent a ubiquitous and highly important motif in many pharmaceutically active compounds and natural products. Development of efficient and straightforward approaches to their enantioselective construction from readily available simple substrates is an important yet challenging goal for synthetic chemistry. Herein we describe an efficient, versatile and facile method for the highly enantioselective construction of tertiary stereocenters via unprecedented consecutive one-pot Suzuki reactions of non-activated racemic 1-chloro-1-iodoalkanes with alkylboranes. It represents the first cross-coupling approach which employs simple and readily available primary alkyl substrates for the Received 16th November 2015 direct multicomponent assembly of enantioenriched tertiary stereocenters. A simple and effective Accepted 6th January 2016 Creative Commons Attribution 3.0 Unported Licence. preparation of 1-chloro-1-iodoalkanes from ubiquitous a-chloroalkanoic acids is also described. DOI: 10.1039/c5sc04378f Collectively, the developed methods open a door to efficient catalytic enantioselective synthesis of www.rsc.org/chemicalscience alkanes bearing tertiary stereocenters from carboxylic acids just in few steps. Introduction (electrophile -

Chirality in Supramolecular Assemblies Chirality in Supramolecular Assemblies
Chirality in Supramolecular Assemblies Chirality in Supramolecular Assemblies Causes and Consequences Edited by F. RICHARD KEENE Department of Chemistry, School of Physical Sciences, University ofAdelaide, Australia WILEY Tbis edition first published 2017 © 2017 John Wiley & Sons, Ltd Registered Office John Wiley & Sons, Ltd, TbeAtrium, Southern Gate, Chichester, West Sussex, P019 8SQ, United Kingdom For details of our global editorial offices, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website atwww.wiley.com. The right of the author to be identified as the author of this work has been asserted in accordance with the Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher. Wiley also publishes its books in a variety of electronic formats. Some content that appears in p1int may not be available in electronic books. Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. The publisher is not associated with any product or vendor mentioned in this book. Limit of Liability/Disclaimer of Warranty: While the publisher and author have used their best efforts in preparing this book, they make no representations or warranties with respect to the accuracy or completeness of the contents of this book and specifically disclaim any implied warranties of merchantability or fitness for a particular purpose. -

Vicinal Dihalide: Two Halogen Atoms Are Bonded to Adjacent Carbons
DAMIETTA UNIVERSITY CHEM-103: BASIC ORGANIC CHEMISTRY LECTURE 4 Dr Ali El-Agamey Types of reactions 1- Addition reaction They normally involves unsaturated compounds capable of accepting additional atoms. 2- Substitution reaction Atom or group replaces atom or group. Types of reactions 3- Elimination reaction Atoms are removed to produce unsaturated compounds or ring. Organic Chemistry, 7th Edition L. G. Wade, Jr. Alkanes © 2010, Prentice Hall Hydrocarbons Hydrocarbons are molecules that are made of carbon and hydrogen ONLY. Alkanes • General formula: CnH2n+2 • Found in everything from natural gas to petroleum. • The smaller alkanes have very low boiling points (b.p.) therefore they are gases. • CH4 C2H6 C3H8 b.p. -160oC -89oC -42oC Preparation of Alkanes • 1- Hydrogenation of alkenes • 2- Alkyl halides (a) Via hydrolysis of Grignard reagent (b) Reduction by metal and acid (c) Coupling with organo-copper compounds. Preparation of Alkanes • (a) Hydrolysis of Grignard reagent Preparation of Alkanes • (b) Reduction by metal and acid Preparation of Alkanes • (c) Coupling with organo-copper compounds. i ' Preparation of Alkanes • (c) Coupling with organo-copper compounds. i Homework (1) Show how can you prepare n-butane from:1 (a) n-butyl bromide (b) sec-butyl bromide and (c) 2-butene. Complete the following equations:1 12 Chapter 8 Reactions of Alkanes • 1- Combustion • 2- Cracking (Pyrolysis) • 3- Halogenation – General reaction; examples – General mechanism; mechanism of specific example (CH4; propane) – Calculation of relative reactivities; -
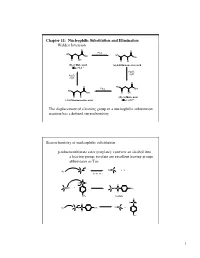
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion O O PCl5 HO HO OH OH O OH O Cl (S)-(-) Malic acid (+)-2-Chlorosuccinic acid [a]D= -2.3 ° Ag2O, H2O Ag2O, H2O O O HO PCl5 OH HO OH O OH O Cl (R)-(+) Malic acid (-)-2-Chlorosuccinic acid [a]D= +2.3 ° The displacement of a leaving group in a nucleophilic substitution reaction has a defined stereochemistry Stereochemistry of nucleophilic substitution p-toluenesulfonate ester (tosylate): converts an alcohol into a leaving group; tosylate are excellent leaving groups. abbreviates as Tos C X Nu C + X- Nu: X= Cl, Br, I O Cl S O O + C OH C O S CH3 O CH3 tosylate O -O S O O Nu C + Nu: C O S CH3 O CH3 1 O Tos-Cl - H3C O O H + TosO - H O H pyridine H O Tos O CH3 [a]D= +33.0 [a]D= +31.1 [a]D= -7.06 HO- HO- O - Tos-Cl H3C O - H O O TosO + H O H pyridine Tos H O CH3 [a]D= -7.0 [a]D= -31.0 [a]D= -33.2 The nucleophilic substitution reaction “inverts” the Stereochemistry of the carbon (electrophile)- Walden inversion Kinetics of nucleophilic substitution Reaction rate: how fast (or slow) reactants are converted into product (kinetics) Reaction rates are dependent upon the concentration of the reactants. (reactions rely on molecular collisions) H H Consider: HO C _ _ C Br Br HO H H H H At a given temperature: If [OH-] is doubled, then the reaction rate may be doubled If [CH3-Br] is doubled, then the reaction rate may be doubled A linear dependence of rate on the concentration of two reactants is called a second-order reaction (molecularity) 2 H H HO C _ _ C Br Br HO H H H H Reaction rates (kinetic) can be expressed mathematically: reaction rate = disappearance of reactants (or appearance of products) For the disappearance of reactants: - rate = k [CH3Br] [OH ] [CH3Br] = CH3Br concentration [OH-] = OH- concentration k= constant (rate constant) L mol•sec For the reaction above, product formation involves a collision between both reactants, thus the rate of the reaction is dependent upon the concentration of both.