I. Isolation of Certain Toxic Components of Kraft Mill Waste and Attempts To
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Retention Indices for Frequently Reported Compounds of Plant Essential Oils
Retention Indices for Frequently Reported Compounds of Plant Essential Oils V. I. Babushok,a) P. J. Linstrom, and I. G. Zenkevichb) National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA (Received 1 August 2011; accepted 27 September 2011; published online 29 November 2011) Gas chromatographic retention indices were evaluated for 505 frequently reported plant essential oil components using a large retention index database. Retention data are presented for three types of commonly used stationary phases: dimethyl silicone (nonpolar), dimethyl sili- cone with 5% phenyl groups (slightly polar), and polyethylene glycol (polar) stationary phases. The evaluations are based on the treatment of multiple measurements with the number of data records ranging from about 5 to 800 per compound. Data analysis was limited to temperature programmed conditions. The data reported include the average and median values of retention index with standard deviations and confidence intervals. VC 2011 by the U.S. Secretary of Commerce on behalf of the United States. All rights reserved. [doi:10.1063/1.3653552] Key words: essential oils; gas chromatography; Kova´ts indices; linear indices; retention indices; identification; flavor; olfaction. CONTENTS 1. Introduction The practical applications of plant essential oils are very 1. Introduction................................ 1 diverse. They are used for the production of food, drugs, per- fumes, aromatherapy, and many other applications.1–4 The 2. Retention Indices ........................... 2 need for identification of essential oil components ranges 3. Retention Data Presentation and Discussion . 2 from product quality control to basic research. The identifi- 4. Summary.................................. 45 cation of unknown compounds remains a complex problem, in spite of great progress made in analytical techniques over 5. -

An Artifact in a Synthetic Pine Oil
RESEARCH NOTE J. Ess. Oil Res., 3, 41-42 (Jan/Feb 1991) An Artifact in a Synthetic Pine Oil Duane F. Zinkel USDA Forest Service, Forest Products Laboratory* One Gifford Pinchot Drive Madison, WI 53705-2398 ABSTRACT: The isopropyl ether of a-terpineol was identified as an artifact in the synthetic pine oil produced when isopropyl alcohol was used as the emulsifier. KEY WORD INDEX: Synthetic pine oil, a-terpineol isopropyl ether, terpinen-4-ol isopropyl ether, turpentine. INTRODUCTION: The manufacture of synthetic pine oil is the primary use for turpentine. The synthesis involves the acid-catalyzed hydration of a-pinene at the in terface ofan emulsion of pinene/mineral acid (1). Various emulsifiers have been used, one of which is isopropyl alcohol. Our gas chromatographic examination of a commercial distilled pine oil, produced using the isopropyl alcohol emulsifier, revealed the presence of 4-5% of a higher boiling component product not present originally in the turpentine. EXPERIMENTAL: NMR spectra were obtained at 310 K with a Bruker WM250 (250 MHz proton and 62.9 MHz carbon) FT spectrometer controlled by an Aspect 2000A minicomputer; DEPT spectra were obtained with a standard Bruker program. Gas chromatography was done with a Hewlett Packard 5880 gas chromatograph (FID) and fused-silica columns: a DB-1 (a methyl silicone) column from J & W Scientific (Folsom, CA), 15m x 0.25mmi.d. witha 0.1-µmfilmoperatedat60°Cand a Carbowaxcolumn,30m x 0.25mm with a 0.25-µm film temperature programmed from 60°C to 225°C at 8°C/min. isopropyl etherwas isolated by liquid chromatography. -

(19) United States (12) Patent Application Publication (10) Pub
US 20060081822A1 (19) United States (12) Patent Application Publication (10) Pub. N0.: US 2006/0081822 A1 Koetzle (43) Pub. Date: Apr. 20, 2006 (54) METHOD TO INCREASE FLASH POINTS OF (52) US. Cl. ............................................................ .. 252/601 FLAMMABLE SOLVENTS (76) Inventor: A. Richard Koetzle, Rochester, NY (57) ABSTRACT (Us) Correspondence AddreSSI The present invention relates to a method to decrease the A- RICHARD KOETZLE ?ammability of normally ?ammable alcohols and solvents. 134 STONECLIFF DRIVE The additive is Alpha Terpineol, Which Will increase the ROCHESTER’ NY 14616 (Us) ?ash point of ?ammable alcohols or solvents, by blending the Terpineol into the ?ammable solvent or alcohol. Solvents (21) Appl' NO" 10/968’441 such as acetone, methanol, ethylacetate, ethanol and Xylene, (22) Filed, Oct 20 2004 to name a feW, increases ?ash points by 50° C. to 60° C., by i ’ addition of 12-14% terpineol. The said solvent can then be Publication Classi?cation blended With other organic solvents to produce performance solvents, such as paint strippers With ?ash points greater (51) Int. Cl. than 1400 F. and meet Federal and state Volatile Organic C 09K 21/00 (2006.01) Compound regulations. US 2006/0081822 A1 Apr. 20, 2006 METHOD TO INCREASE FLASH POINTS OF [0008] The organic solvent or combination of solvents can FLAMMABLE SOLVENTS comprise up to 99 Weight percent of the composition in total, and may be the combination of tWo or more different types BACKGROUND OF THE INVENTION of organic solvents. A typical combination may comprise; [0001] Many industrial processing cleaning compositions [0009] 1.0 to 99 Weight percent organic solvent. -

Immobilization of P. Digitatum and Bioconversion of Limonene to Alpha-Terpineol. Qiang Tan Louisiana State University and Agricultural & Mechanical College
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1996 Immobilization of P. Digitatum and Bioconversion of Limonene to Alpha-Terpineol. Qiang Tan Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Tan, Qiang, "Immobilization of P. Digitatum and Bioconversion of Limonene to Alpha-Terpineol." (1996). LSU Historical Dissertations and Theses. 6312. https://digitalcommons.lsu.edu/gradschool_disstheses/6312 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -

Wöhler Synthesis of Urea
Wöhler synthesis of urea Wöhler, 1928 – – + + NH Cl + K N C O 4 NH4 N C O O H2N NH2 Friedrich Wöhler Annalen der Physik und Chemie 1828, 88, 253–256 Significance: Wöhler was the first to make an organic substance from an inorganic substance. This was the beginning of the end of the theory of vitalism: the idea that organic and inorganic materials differed essentially by the presence of the “vital force”– present only in organic material. To his mentor Berzelius:“I can make urea without thereby needing to have kidneys, or anyhow, an animal, be it human or dog" Friedrich Wöhler, 1880-1882 Polytechnic School in Berlin Support for vitalism remained until 1845, when Kolbe synthesized acetic acid Polytechnic School at Kassel from carbon disulfide University of Göttingen Fischer synthesis of glucose PhHN N O N O NHPh Br aq. HCl Δ PhNH2NH2 HO H HO H O Br "α−acrose" H OH H OH H OH H OH CH2OH CH2OH α−acrosazone α−acrosone an “osazone” (osazone test for reducing sugars) Zn/AcOH OH OH CO2H CHO O HO H HO H OH Br2 HO H HO H Na-Hg HO H HNO3 HO H H OH H OH H OH H OH then resolve H O+ H OH via strychnine H OH H OH 3 H OH CH2OH salts CH2OH CH OH CH2OH 2 D-Mannonic acid DL-Mannose DL-Mannitol DL-Fructose quinoline • Established stereochemical relationship CO2H CHO H OH H OH between mannose and glucose Na-Hg HO H HO H (part of Fischer proof) H OH + H OH H3O • work mechanisms from acrosazone on H OH H OH Emil Fischer, 1852-1919 CH2OH CH2OH University of Munich (1875-81) D-Gluconic acid D-Glucose University of Erlangen (1881-88) University of Würzburg (1888-92) University of Berlin (1892-1919) Fischer, E. -
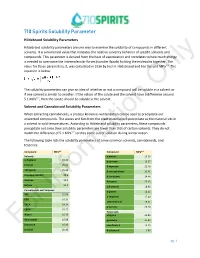
710 Spirits Solubility Parameter Hildebrand Solubility Parameters
710 Spirits Solubility Parameter Hildebrand Solubility Parameters Hildebrand solubility parameters are one way to examine the solubility of compounds in different solvents. It is a numerical value that indicates the relative solvency behavior of specific solvents and compounds. This parameter is derived from the heat of vaporization and correlates to how much energy is needed to overcome the intermolecular forces (van der Waals) holding the molecules together. The value for these parameters, δ, was calculated in 1936 by Joel H. Hildebrand and has the unit MPa1/2. The equation is below: The solubility parameters can give an idea of whether or not a compound will be soluble inOnly a solvent or if one solvent is similar to another. If the values of the solute and the solvent have a difference around 5.1 MPa1/2, then the solute should be soluble in the solvent. Solvent and Cannabinoid Solubility Parameters When extracting cannabinoids, a process known as winterization can be used to precipitate out unwanted compounds. The waxes and fats from the plant material will precipitate as the material sits in a solvent in cold temperatures. According to Hildebrand solubility parameters, these compounds precipitate out since their solubility parameters are lower than that of certain solvents. They do not match the difference of 5.1 MPa1/2 so they come out of solution during winterization. The following table lists the solubility parameters of some common solvents, cannabinoids, and terpenes: Compound MPa1/2 Compound MPa1/2 Solvents a-pinene 16.16 n-Heptane 15.40 p-cymene 16.57 Ethanol 26.20 B-myrcene 21.46 710 Spirits 25.66 B-caryophyllene 21.41 Isopropyl Alcohol 23.6 d-Limonene 24.46 Acetone 19.9 Pulegone 25.65 Butane 14.1 1,8-cineole 18.96 Cannabinoids and Terpenes a-pinene 16.15 THC Information22.09 a-terpineol 17.10 CBD 24.34 terpineol-4-ol 18.21 THCA 24.14 p-cymene 24.20 CBDA 25.77 Flavonoids Waxes 16.36 apigenin 20.80 Chlorophyll 16.98 quercetin 25.89 Limonene 16.90 cannflavin A 10.35 ForLinalool 20.46 B-sitosterol 3.86 pg. -

Flavonoids and Terpenes • Analytical Standards for Terpenes and Flavonoid Analysis • Single-Element Standards and Mixes Available
cannstandards® Flavonoids and Terpenes • Analytical standards for terpenes and flavonoid analysis • Single-Element Standards and Mixes Available spexcertiprep.com Connect with us Phone: +1.732.549.7144 • +1.800.LAB.SPEX Spex CertiPrep is an Fax: +1.732.603.9647 Anatylia Scientific company [email protected] Find out more at antylia.com Flavonoids and Terpenes Flavonoids are naturally occurring secondary metabolic products which can have important functions within plants and benefit consumers with health and healing properties. Organic Certified Many beneficial compounds are metabolites produced as an end product Reference Materials of chemical and biological processes. Metabolites are small molecules that have many functions including defense, pigments, pheromones, odorants and catalysts. Primary metabolites are necessary for growth, development and reproduction. Flavonoids are secondary plant, algae or fungus metabolites composed of polyphenolic compounds. Secondary metabolites are not directly involved in critical processes but have secondary functions involving defense and pigmentation. We offer analytical standards for flavonoid analysis. Analytical Standards for Terpenes are the common term for a large group of compounds that contribute Flavonoid and Terpene Testing to flavor and smell of botanical products. Custom standards are also available. Contact us at 732.549.7144 or via email at [email protected] to discuss your specific requirements. Supplied with a Certificate of Analysis to 1702 ed 5 & it 1 d 7 re 0 c 3 c 4 A C er -

Tea Tree Oil
CONTACT DERMATITIS Tea Tree Oil Orville Hartford, MD; Kathryn A. Zug, MD Tea tree oil is a popular ingredient in many over- more than 15% 1,8-cineole (eucalyptol).3,4 Although the-counter healthcare and cosmetic products. eucalyptol is not an irritant, eucalyptus essential oil With the explosion of the natural and alternative has eucalyptol as its major component.2 medicine industry, more and more people are using products containing tea tree oil. This arti- Sources and Exposure cle reviews basic information about tea tree oil Mass marketing of Australian tea tree oil as a nat- and contact allergy, including sources of tea tree ural cure for a variety of skin conditions has lead to oil, chemical composition, potential cross reac- its inclusion in products such as cosmetics, sham- tions, reported cases of allergic contact dermati- poos, mouthwashes, ointments, soaps, lotions, tis, allergenic compounds in tea tree oil, deodorants, sunscreens, laundry detergents, tooth- practical patch testing information, and preven- paste, fabric softeners, and cleansers.1 Tea tree oil tive measures. also is used for massage and aromatherapy and can Cutis. 2005;76:178-180. be found in nonprescription medications for the treatment of athlete’s foot, warts, acne, bacterial infections, lice, and psoriasis.5 Finally, tea tree oil is used by the general public and paramedical practi- Allergen Aspects tioners for a myriad of conditions.6 Tea tree oil is an essential oil most often distilled Tea tree oil’s topical antimicrobial activity has from the terminal branches -

(CDR) by CASRN Or Accession Number
List of Chemicals Reported for the 2012 Chemical Data Reporting (CDR) by CASRN or Accession Number For the 2012 CDR, 7,674 unique chemicals were reported by manufacturers (including importers). Chemicals are listed by CAS Registry Number (for non-confidential chemicals) or by TSCA Accession Number (for chemicals listed on the confidential portion of the TSCA Inventory). CASRN or CASRN or ACCESSION ACCESSION NUMBER CA INDEX NAME or GENERIC NAME NUMBER CA INDEX NAME or GENERIC NAME 100016 Benzenamine, 4-nitro- 10042769 Nitric acid, strontium salt (2:1) 10006287 Silicic acid (H2SiO3), potassium salt (1:2) 10043013 Sulfuric acid, aluminum salt (3:2) 1000824 Urea, N-(hydroxymethyl)- 10043115 Boron nitride (BN) 100107 Benzaldehyde, 4-(dimethylamino)- 10043353 Boric acid (H3BO3) 1001354728 4-Octanol, 3-amino- 10043524 Calcium chloride (CaCl2) 100174 Benzene, 1-methoxy-4-nitro- 100436 Pyridine, 4-ethenyl- 10017568 Ethanol, 2,2',2''-nitrilotris-, phosphate (1:?) 10043842 Phosphinic acid, manganese(2+) salt (2:1) 2,7-Anthracenedisulfonic acid, 9,10-dihydro- 100447 Benzene, (chloromethyl)- 10017591 9,10-dioxo-, sodium salt (1:?) 10045951 Nitric acid, neodymium(3+) salt (3:1) 100185 Benzene, 1,4-bis(1-methylethyl)- 100469 Benzenemethanamine 100209 1,4-Benzenedicarbonyl dichloride 100470 Benzonitrile 100210 1,4-Benzenedicarboxylic acid 100481 4-Pyridinecarbonitrile 10022318 Nitric acid, barium salt (2:1) 10048983 Phosphoric acid, barium salt (1:1) 9-Octadecenoic acid (9Z)-, 2-methylpropyl 10049044 Chlorine oxide (ClO2) 10024472 ester Phosphoric acid, -

Chemical Resistance Chart
A = Good Resistance | B = Fair Resistance | C = Depends on Conditions | D = Not Recommended Buna/Nitrile EPDM FKM Silicone PTFE* Butyl** Buna/Nitrile EPDM FKM Silicone PTFE* Butyl** Acetal D B D – A B Amyl Acetone D B D – A B Acetaldehyde D A D B A A Amyl Alcohol A A A D A A Acetamide A A B – A A Amyl Borate A D A – A D Acetate Solvents D A D – A C Amyl Chloride D D A D A D Acetic Acid, 10% B A C – A B Amyl Chloronapthalene D D A – A D Acetic Acid, 30% D A C – A B Amyl Napthalene D D A – A D Acetic Acid, 50% D A D – A B Amyl Oleate D B C – A B Acetic Acid, Glacial D A D – A B Amyl Phenol D D A – A D Acetic Anhydride D B D D A B Anethole D D B – A D Acetic Ester (Ethyl Acetate) D A D – A B Aniline D D B B B B Acetic Ether (Ethyl Acetate) D A D – A B Aniline Dyes D B B – A B Acetic Oxide (Acetic Anhydride) D B D – A B Aniline Hydrochloride B B B – A B Acetone D A D D A B Animal Fats A C A – A C Acetophenome D A D – A A Animal Grease A C A – A D Acetyl Acetone C B D – A B Animal Oils A C A – A C Acetyl Chloride D C B C A C Ansul Ether D C D – A D Acetylene A B A – A A Antifreeze A A A – A A Acrylonitrile D D D – A D Antimony Chloride A D A – A B Air A A A A A A Antimony Pentachloride B D A – A D Alcohol Aliphatic A A C – B A Aqua Regia D B A – A C Alcohol, Aromatic C D A – A D Aromatic Hydrocarbons D D A – A D Alk-Tri (Trichloroethylene) D D A – A D Arquad A A A – A A Allyl Alcohol A A B – A A Arsenic Acid A A A – A A Allyl Bromide D D B – A D Arsenic Chloride C D D – A D Allyl Chloride D D A – A D Arsenic Trichloride A D D – A D Alum (Alum -

US2566956.Pdf
Patented Sept. 4, 1951 ... ??? .. 2,566,956 UNITED STATES PATENT office George Wesley Pedlow, Jr. and Carl Shelley. Miner, Jr., Evanston, Ill., assignors to Minne sota Mining & Manufacturing Company, St. Paul, Minn., a corporation of Delaware Application December 31, 1942, Serial No. 40,904 1 Claim. (Cl. 260-4488) 1. The present invention relates to organic silicon It will be noted that this procedure does not yield Compounds and to products or articles involving 'an alkoxy silicane, nor an alkoxy chloro sili the same, and to methods for producing such cane; rather it yields an alkyl silicane or an alkyl Compounds. chloro silicane, such as: - Certain of the organic silicon compounds in s C question may be regarded as silicon tetrachloride, \sic, SiCl4 (which is derivable from silica, SiO2), in which at least one of the chlorine atoms is re ' ... - ??” placed by an organic radical, particularly an in which the carbon atom of the alkyl group alkoxy radical, yielding, compounds such as (or of an aryl group) is joined directly to the sili ... (RO)2SiCl2 and where the compounds in ques con atom; whereas in alkoxy silicon compounds tion have certain special properties and novel there is an oxygen atom intervening between the '-characteristics and/or are adapted for new and carbon atom of the alkyl group and the silicon valuable uses. The invention as a Whole will atom, which materially alters the nature of the be more fully explained hereinafter. 5 product:: - Further, the Grignard reaction is very Heretofore others have produced certain or expensive and hence unsuitable for ordinary large ganic silicon containing products, starting with scale commercial operations, at least in many silicon tetrachloride, SiCl4. -

Multi-Statistical Approach for the Study of Volatile Compounds of Industrial Spoiled Manzanilla Spanish-Style Table Olive Fermentations
foods Article Multi-Statistical Approach for the Study of Volatile Compounds of Industrial Spoiled Manzanilla Spanish-Style Table Olive Fermentations Antonio Garrido-Fernández, Alfredo Montaño, Amparo Cortés-Delgado, Francisco Rodríguez-Gómez and Francisco Noé Arroyo-López * Food Biotechnology Department, Instituto de la Grasa (CSIC), University Pablo de Olavide, Building 46, Crta, Utrera km 1, 41013 Seville, Spain; [email protected] (A.G.-F.); [email protected] (A.M.); [email protected] (A.C.-D.); [email protected] (F.R.-G.) * Correspondence: [email protected] Abstract: Table olives can suffer different types of spoilage during fermentation. In this work, a multi-statistical approach (standard and compositional data analysis) was used for the study of the volatile organic compounds (VOCs) associated with altered (butyric, sulfidic, and putrid) and non- altered (normal) Manzanilla Spanish-style table olive fermentations. Samples were collected from two industrial fermentation yards in Seville (Spain) in the 2019/2020 season. The VOC profiles of altered (n = 4) and non-altered (n = 6) samples were obtained by headspace solid-phase microextraction combined with gas chromatography-mass spectrometry (HS-SPME-GC-MS). Ninety-one VOCs were identified and grouped into alcohols (30), esters (21), carbonyl compounds (12), acids (10), terpenes Citation: Garrido-Fernández, A.; (6), phenols (6), sulfur compounds (2), and others (4). The association of the VOCs with spoilage Montaño, A.; Cortés-Delgado, A.; samples depended on the standard or compositional statistical methodology used. However, butyric Rodríguez-Gómez, F.; Arroyo-López, spoilage was strongly linked by several techniques to methyl butanoate, ethyl butanoate, and butanoic F.N.