Product Monograph
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
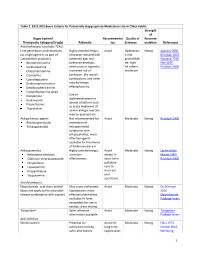
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

Arrhythmias in the Intensive Care Patient Hans-Joachim Trappe, Bodo Brandts and Peter Weismueller
Arrhythmias in the intensive care patient Hans-Joachim Trappe, Bodo Brandts and Peter Weismueller Purpose of review ventricular fibrillation. The use of automatic external Atrial fibrillation, atrial flutter, AV–nodal reentry tachycardia defibrillators by basic life support ambulance providers or first with rapid ventricular response, atrial ectopic tachycardia, and responder in early defibrillation programs has been associated preexcitation syndromes combined with atrial fibrillation or with a significant increase in survival rates. Drugs such as ventricular tachyarrhythmias are typical arrhythmias in intensive lidocaine, procainamide, sotalol, amiodarone, or magnesium care patients. Most frequently, the diagnosis of the underlying were recommended for treatment of ventricular arrhythmia is possible from the physical examination, the tachyarrhythmias in intensive care patients. Amiodarone is a response to maneuvers or drugs, and the 12-lead surface highly efficacious antiarrhythmic agent for many cardiac electrocardiogram. In all patients with unstable hemodynamics, arrhythmias, ranging from atrial fibrillation to malignant immediate DC-cardioversion is indicated. Conversion of atrial ventricular tachyarrhythmias, and seems to be superior to other fibrillation to sinus rhythm is possible using antiarrhythmic antiarrhythmic agents. drugs. Amiodarone has a conversion rate in atrial fibrillation of up to 80%. However, caution in the use of short-term Keywords administration of intravenous amiodarone in critically ill patients atrial fibrillation, -

Safety and Efficacy of Ibutilide in Cardioversion of Atrial Flutter And
J Am Board Fam Med: first published as 10.3122/jabfm.2011.01.080096 on 5 January 2011. Downloaded from CLINICAL REVIEW Safety and Efficacy of Ibutilide in Cardioversion of Atrial Flutter and Fibrillation Madhuri Nair, MD, Lekha K. George, MD, and Santhosh K. G. Koshy, MD This article reviews the safety and efficacy of ibutilide for use in patients with atrial fibrillation and flut- ter. Ibutilide, a class III antiarrhythmic agent, is primarily used for conversion of atrial flutter and fi- brillation and is a good alternative to electrical cardioversion. Ibutilide has a conversion rate of up to 75% to 80% in recent-onset atrial fibrillation and flutter; the conversion rate is higher for atrial flutter than for atrial fibrillation. It is also safe in the conversion of chronic atrial fibrillation/flutter among patients receiving oral amiodarone therapy. Ibutilide pretreatment facilitates transthoracic defibrilla- tion and decreases the energy requirement of electrical cardioversion by both monophasic and biphasic shocks. Pretreatment with ibutilide before electrical defibrillation has a conversion rate of 100% com- pared with 72% with no pretreatment. Ibutilide is also safe and efficient in the treatment of atrial fibril- lation in patients who have had cardiac surgery, and in accessory pathway–mediated atrial fibrillation where the conversion rate of ibutilide is as high as 95%. There is up to a 4% risk of torsade de pointes and a 4.9% risk of monomorphic ventricular tachycardia. Hence, close monitoring in an intensive care unit setting is warranted during and at least for 4 hours after drug infusion. The anticoagulation strat- egy is the same as for any other mode of cardioversion.(J Am Board Fam Med 2011;24:86–92.) Keywords: Antiarrhythmics, Arrhythmia, Atrial Fibrillation, Cardiovascular Disorders, Cardioversion, Drug Ther- copyright. -

Efficacy and Safety of Ibutilide for the Cardioversion of Atrial Fibrillation: a Systematic Review and Meta- Analysis
Open Access Austin Surgery Case Reports Review Article Efficacy and Safety of Ibutilide for the Cardioversion of Atrial Fibrillation: A Systematic Review and Meta- Analysis Gong CC1, Tang Y2, Huang Y2 and Liu X2* 1Zhejiang University School of Medicine Children’s Abstract Hospital, China Background: Ibutilide has been approved for cardioversion of Atrial 2Department of Critical Care Medicine, the Affiliated Fibrillation (AF), but its side-effects include a high risk of torsade de pointes, Hospital of Guizhou Medical University, China besides, one recent meta-analysis showed ibutilide was inferior to vernakalant *Corresponding author: Liu X, Department of Critical for conversion (AF<7 days). Hence, the aim of this study is to evaluate the Care Medicine, the Affiliated Hospital of Guizhou Medical efficacy and safety of ibutilide for the cardioversion of AF within 90 days. University, China Methods: The Embase, PubMed, Web of Science, Cochrane Central Received: February 16, 2021; Accepted: March 17, databases and clinical trials.gov were comprehensively searched for relevant 2021; Published: March 24, 2021 studies from January 1991 to May 2020 using the keywords “ibutilide” and “atrial fibrillation”. Only Randomized Controlled Trials (RCTs) comparing ibutilide with placebo or other Anti-Arrhythmic Drugs (AADs) for the termination of AF (duration of AF ≤90 days) were included. The primary outcome was successful cardioversion in response to ibutilide versus placebo or other AADs within 4h. Related adverse events were defined as secondary outcomes. Results: A total of 1712 patients in 13 RCTs met the eligibility criteria. Four trials compared ibutilide to placebo; nine trials compared ibutilide to other active drugs. -

Keeping up with the Pace of Antiarrhythmic Drugs ANNMARIE PALATNIK, APN,BC, MSN Coordinator of Continuing Education • Virtua Health • Marlton, N.J
D rug File Keeping up with the pace of antiarrhythmic drugs ANNMARIE PALATNIK, APN,BC, MSN Coordinator of Continuing Education • Virtua Health • Marlton, N.J. HAVE YOU NOTICED how challenging it is to Conducting impulses keep pace with the The conduction system of the heart, shown below, begins with the heart’s natural pacemaker, the changing beat of phar- sinoatrial (SA) node. When an impulse leaves the SA node, it travels through the atria along macology? Just as you Bachmann’s bundle and the internodal pathways on its way to the atrioventricular (AV) node. After learn the latest drugs the impulse passes through the AV node, it travels to the ventricles, first down the bundle of His, and classifications, new then along the bundle branches and, finally, down the Purkinje fibers. classes are developed and new drugs added to Bachmann’s bundle classes. Even drugs that have been on the mar- ket a long time can have dosing and indica- SA node tion changes. Antiarrhythmic Internodal tracts drugs, which restore Posterior (Thorel’s) normal rhythm and Middle (Wenckebach’s) conduction to the heart, Anterior are no exception. In this AV node article, I’ll bring you up-to-date on the Bundle of His antiarrhythmics now Right bundle branch available. Left bundle branch Class assignments Most antiarrhythmic drugs used to slow a rapid heart rate are classified according to the Vaughn Williams classification system Purkinje fibers (see Classifying Anti- arrhythmics the Vaughn Williams way). These drugs fall Class I into four general groups in this classification system Sodium channel blockers stop the flow of sodium into with each group having several subgroups. -

Ventricular Tachycardia Drugs Versus Devices John Camm St
Cardiology Update 2015 Davos, Switzerland: 8-12th February 2015 Ventricular Arrhythmias Ventricular Tachycardia Drugs versus Devices John Camm St. George’s University of London, UK Imperial College, London, UK Declaration of Interests Chairman: NICE Guidelines on AF, 2006; ESC Guidelines on Atrial Fibrillation, 2010 and Update, 2012; ACC/AHA/ESC Guidelines on VAs and SCD; 2006; NICE Guidelines on ACS and NSTEMI, 2012; NICE Guidelines on heart failure, 2008; NICE Guidelines on Atrial Fibrillation, 2006; ESC VA and SCD Guidelines, 2015 Steering Committees: multiple trials including novel anticoagulants DSMBs: multiple trials including BEAUTIFUL, SHIFT, SIGNIFY, AVERROES, CASTLE- AF, STAR-AF II, INOVATE, and others Events Committees: one trial of novel oral anticoagulants and multiple trials of miscellaneous agents with CV adverse effects Editorial Role: Editor-in-Chief, EP-Europace and Clinical Cardiology; Editor, European Textbook of Cardiology, European Heart Journal, Electrophysiology of the Heart, and Evidence Based Cardiology Consultant/Advisor/Speaker: Astellas, Astra Zeneca, ChanRX, Gilead, Merck, Menarini, Otsuka, Sanofi, Servier, Xention, Bayer, Boehringer Ingelheim, Bristol- Myers Squibb, Daiichi Sankyo, Pfizer, Boston Scientific, Biotronik, Medtronic, St. Jude Medical, Actelion, GlaxoSmithKline, InfoBionic, Incarda, Johnson and Johnson, Mitsubishi, Novartis, Takeda Therapy for Ventricular Tachycardia Medical therapy Antiarrhythmic drugs Autonomic management Ventricular tachycardia Monomorphic Polymorphic Ventricular fibrillation Ventricular storms Ablation therapy Device therapy Surgical Defibrillation Catheter Antitachycardia pacing History of Antiarrhythmic Drugs 1914 - Quinidine 1950 - Lidocaine 1951 - Procainamide 1946 – Digitalis 1956 – Ajmaline 1962 - Verapamil 1962 – Disopyramide 1964 - Propranolol 1967 – Amiodarone 1965 – Bretylium 1972 – Mexiletine 1973 – Aprindine, Tocainide 1969 - Diltiazem 1975- Flecainide 1976 – Propafenone Encainide Ethmozine 2000 - Sotalol D-sotalol 1995 - Ibutilide (US) Recainam 2000 – Dofetilide US) IndecainideX Etc. -

Superiority of Ibutilide (A New Class III Agent) Over
568 Heart 1998;79:568–575 Superiority of ibutilide (a new class III agent) over DL-sotalol in converting atrial flutter and atrial Heart: first published as 10.1136/hrt.79.6.568 on 1 June 1998. Downloaded from fibrillation M A Vos, S R Golitsyn, K Stangl, M Y Ruda, L Van Wijk, J D Harry, K T Perry, P Touboul, G Steinbeck, HJJWellens, for the Ibutilide/Sotalol Comparator Study Group Abstract III drug, under monitored conditions, is a Objective—To compare the eYcacy and potential alternative to currently available safety of a single dose of ibutilide, a new cardioversion options. class III antiarrhythmic drug, with that of (Heart 1998;79:568–575) DL-sotalol in terminating chronic atrial fibrillation or flutter in haemodynami- Keywords: atrial fibrillation; atrial flutter; cally stable patients. antiarrhythmic agents; ibutilide; sotalol Design—Double blind, randomised study. Setting—43 European hospitals. Atrial fibrillation and atrial flutter are the most Patients—308 patients (mean age 60 years, 70% men, 48% with heart disease) with frequently occurring tachycardias in man: sustained atrial fibrillation (n = 251) or prevalence is estimated to be 0.4% for atrial 1–3 atrial flutter (n = 57) (duration three fibrillation and 0.1% for atrial flutter, and hours to 45 days) were randomised to they may increase to 4% in people older than University Hospital, three groups to receive a 10 minute 60 years. Treatment for acute conversion of Maastricht, infusion of 1 mg ibutilide (n = 99), 2 mg these arrhythmias consists of antiarrhythmic Netherlands drugs or electrical cardioversion, or both. MAVos ibutilide (n = 106), or 1.5 mg/kg DL-sotalol HJJWellens (n = 103). -

FDA Briefing Document Cardiovascular and Renal Drugs
FDA Briefing Document Cardiovascular and Renal Drugs Advisory Committee (CRDAC) Meeting December 10, 2019 Topic: New Drug Application 22034 Vernakalant Hydrochloride Injection for the Rapid Conversion of Recent Onset Atrial Fibrillation 1 The attached package contains background information prepared by the Food and Drug Administration (FDA) for the panel members of the advisory committee. The FDA background package often contains assessments and/or conclusions and recommendations written by individual FDA reviewers. Such conclusions and recommendations do not necessarily represent the final position of the individual reviewers, nor do they necessarily represent the final position of the Review Division or Office. We have brought New Drug Application 22034, atrial fibrillation for the treatment of recent onset atrial fibrillation, to this Advisory Committee in order to gain the Committee’s insights and opinions on key issues identified by the Agency. The background package may not include all issues relevant to the final regulatory recommendation, and the final determination may be affected by issues not discussed at the advisory committee meeting. The FDA will not issue a final determination on the issues at hand until input from the advisory committee process has been considered and all reviews have been finalized. 2 Table of Contents Glossary .......................................................................................................................................... 8 1. Introduction .......................................................................................................................... -

PHARMACY TIMES by IEHP PHARMACEUTICAL SERVICES DEPARTMENT February 11, 2013
PHARMACY TIMES BY IEHP PHARMACEUTICAL SERVICES DEPARTMENT February 11, 2013 The Centers for Medicare and Medicaid Services (CMS) developed performance and quality measures to help Medicare beneficiaries make informed decisions regarding health and prescription drug plans. As part of this effort, CMS adopted measures for High Risk Medication (HRM) endorsed by the Pharmacy Quality Alliance (PQA) and the National Quality Forum (NQF). The HRM was developed using existing HEDIS measurement “Drugs to be avoided in the elderly”. The HRM rate analyzes the percentage of Medicare Part D beneficiaries 65 years or older who have received prescriptions for drugs with a high risk of serious side effects in the elderly. In order to advance patient safety, IEHP will be identifying members over 65 and currently on one of the medications identified in Table 1. Providers will be receiving a list of these members from IEHP on an ongoing basis. IEHP asks providers to review their member’s current drug regimen and safety risk and make any appropriate changes when applicable. Table 1: Medications identified by CMS to be high risk in the elderly: Drug Class Drug Safety Concerns IEHP Formulary Alternative(s) Acetylcholinesterase Donepezil (in patients Orthostatic hypotension or Memantine Inhibitor with syncope) bradycardia Amphetamines Dextroamphetamine CNS stimulation Weight Control: Diet Lisdextroamphetamine & lifestyle Diethylpropion modification Methylphenidate Phentermine Depression: mirtazapine, trazodone Analgesic Pentazocine Confusion, hallucination, Mild Pain: (includes Meperidine delirium, fall, fracture APAP combination Tramadol Lowers seizure threshold medications) Aspirin > 325 mg/day GI bleeding/peptic ulcer, edema Mod-Severe Pain Diflunisal may worsen heart failure Norco Etodolac Vicodin Fenoprofen Percocet Ketoprofen Morphine Meclofenamate Mefenamic acid Nabumetone 303 E. -

FDA Listing of Established Pharmacologic Class Text Phrases January 2021
FDA Listing of Established Pharmacologic Class Text Phrases January 2021 FDA EPC Text Phrase PLR regulations require that the following statement is included in the Highlights Indications and Usage heading if a drug is a member of an EPC [see 21 CFR 201.57(a)(6)]: “(Drug) is a (FDA EPC Text Phrase) indicated for Active Moiety Name [indication(s)].” For each listed active moiety, the associated FDA EPC text phrase is included in this document. For more information about how FDA determines the EPC Text Phrase, see the 2009 "Determining EPC for Use in the Highlights" guidance and 2013 "Determining EPC for Use in the Highlights" MAPP 7400.13. -

Comparison of the Effects of DC031050, a Class III Antiarrhythmic Agent, on Herg Channel and Three Neuronal Potassium Channels
npg Acta Pharmacologica Sinica (2012) 33: 728–736 © 2012 CPS and SIMM All rights reserved 1671-4083/12 $32.00 www.nature.com/aps Original Article Comparison of the effects of DC031050, a class III antiarrhythmic agent, on hERG channel and three neuronal potassium channels Ping LI1, Hai-feng SUN1, Ping-zheng ZHOU1, Chao-ying MA2, Guo-yuan HU1, Hua-liang JIANG1, Min LI1, Hong LIU1, *, Zhao- bing GAO1, * 1State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China; 2Life Science and Engineering College of Southwest Jiaotong University, Chengdu 610031, China Aim: This study was conducted to test the selectivity of DC031050 on cardiac and neuronal potassium channels. Methods: Human ether-à-go-go related gene (hERG), KCNQ and Kv1.2 channels were expressed in CHO cells. The delayed rectifier potassium current (IK) was recorded from dissociated hippocampal pyramidal neurons of neonatal rats. Whole-cell voltage patch clamp was used to record the voltage-activated potassium currents. Drug-containing solution was delivered using a RSC-100 Rapid Solution Changer. Results: Both DC031050 and dofetilide potently inhibited hERG currents with IC50 values of 2.3±1.0 and 17.9±1.2 nmol/L, respectively. DC031050 inhibited the IK current with an IC50 value of 2.7±1.5 μmol/L, which was >1000 times the concentration required to inhibit hERG current. DC031050 at 3 μmol/L did not significantly affect the voltage-dependence of the steady activation, steady inactivation of IK, or the rate of IK from inactivation. Intracellular application of DC031050 (5 μmol/L) was insufficient to inhibit IK. -

First Case of Acute Poisoning with Amiodarone and Flecainide in Attempted Suicide Successfully Managed with Lipid Emulsion Thera
healthcare Case Report First Case of Acute Poisoning with Amiodarone and Flecainide in Attempted Suicide Successfully Managed with Lipid Emulsion Therapy in the Emergency Department: Case Report and Literature Review Cristina Bologa 1,2, Catalina Lionte 1,2,* , Alexandra Popescu 2, Victorita Sorodoc 1,2 and Laurentiu Sorodoc 1,2 1 Internal Medicine and Clinical Toxicology Department, “Grigore T. Popa” University of Medicine and Pharmacy, 700115 Iasi, Romania; cristina.bologa@umfiasi.ro (C.B.); victorita.sorodoc@umfiasi.ro (V.S.); laurentiu.sorodoc@umfiasi.ro (L.S.) 2 2nd Medical Clinic, “Sf. Spiridon” Emergency Clinical County Hospital, 700111 Iasi, Romania; [email protected] * Correspondence: catalina.lionte@umfiasi.ro or [email protected]; Tel.: +40-742082318 Abstract: Acute antiarrhythmics poisoning represents a challenge in the Emergency Department (ED). These patients often develop malignant arrhythmias in need of exceptional therapeutic measures in the ICU. We report a 47-year-old patient admitted to the ED 5 h after the ingestion of a large dose of amiodarone and flecainide in a suicide attempt. During their ED stay, the patient developed signs of cardiotoxicity evidenced by electrocardiogram and ventricular arrhythmias. The toxicological results showed a level of 4.8 mg/L amiodarone and 2.98 mg/L flecainide. He was successfully treated in the Citation: Bologa, C.; Lionte, C.; ED using a large dose of sodium bicarbonate and lipid emulsion therapy. After hospital admission, Popescu, A.; Sorodoc, V.; Sorodoc, L. First Case of Acute Poisoning with he remained stable, with no need for exceptional therapeutic measures such as mechanical circulatory Amiodarone and Flecainide in support, cardiac pacing or ECMO.