Rapid Host Immune Response and Viral Dynamics in Herpes Simplex Virus-2 Infection
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Newer Trends in the Management of Genital Herpes
Review NNewerewer trendstrends inin thethe mmanagementanagement ofof genitalgenital herpesherpes Article AAmiyamiya KKumarumar NNath,ath, DDevinderevinder MohanMohan ThappaThappa Department of Dermatology ABSTRACT and STD, Jawaharlal Institute of Postgraduate Medical Management of genital herpes is complex. Apart from using the standard antivirals, an Education and Research (JIPMER), Pondicherry - 605 ideal management protocol also needs to address various aspects of the disease, including 006, India the psychological morbidity. Oral acyclovir, valacyclovir or famciclovir are recommended for routine use. Long-term suppressive therapy is effective in reducing the number of AAddressddress forfor ccorrespondence:orrespondence: recurrences and the risk of transmission to others. Severe or disseminated disease may Dr. Devinder Mohan Thappa, require intravenous therapy. Resistant cases are managed with foscarnet or cidofovir. Genital Department of Dermatology herpes in human immunodeÞ ciency virus-infected individuals usually needs a longer duration and STD, JIPMER, of antiviral therapy along with continuation of highly active anti retroviral therapy (HAART). Pondicherry - 605 006, India. E-mail: [email protected] Genital herpes in late pregnancy increases the risk of neonatal herpes. Antiviral therapy and/or cesarean delivery are indicated depending on the clinical circumstance. Acyclovir appears to be safe in pregnancy. But, there is limited data regarding the use of valacyclovir and famciclovir in pregnancy. Neonatal herpes requires a higher dose of acyclovir given intravenously for a longer duration. Management of the sex partner, counseling and prevention advice are equally important in appropriate management of genital herpes. Vaccines till date have been marginally effective. Helicase–primase inhibitors, needle-free mucosal vaccine and a new microbicide product named VivaGel may become promising treatment options in the future. -

Guide for Common Viral Diseases of Animals in Louisiana
Sampling and Testing Guide for Common Viral Diseases of Animals in Louisiana Please click on the species of interest: Cattle Deer and Small Ruminants The Louisiana Animal Swine Disease Diagnostic Horses Laboratory Dogs A service unit of the LSU School of Veterinary Medicine Adapted from Murphy, F.A., et al, Veterinary Virology, 3rd ed. Cats Academic Press, 1999. Compiled by Rob Poston Multi-species: Rabiesvirus DCN LADDL Guide for Common Viral Diseases v. B2 1 Cattle Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 2 Deer and Small Ruminants Please click on the principle system involvement Generalized viral disease Respiratory viral disease Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 3 Swine Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 4 Horses Please click on the principle system involvement Generalized viral diseases Neurological viral diseases Respiratory viral diseases Enteric viral diseases Abortifacient/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 5 Dogs Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. -

Pt Education-Rubella
Patient Education Healthcare Epidemiology and Infection Control Rubella An illness requiring droplet precautions This handout describes What is Rubella? Rubella and it symptoms. It Rubella (also called German measles, 3-day measles or Rubella virus also explains how this infection) is a viral disease. It can be prevented with a vaccine. disease can be spread and You are not at risk if you have: offers steps to prevent • Had blood tests showing that you are immune due to a history of others from getting it. clinical disease. To learn more about Rubella, • Received 2 doses of the MMR (Mumps, Measles, Rubella) vaccine. visit these Web sites: Rubella is a reportable disease. The health department is notified when a case is diagnosed to protect others who may have come in contact with www.cdc.gov/ncidod/dvrd/ you and are at risk of becoming ill. revb/measles/rubella_index The greatest danger from rubella is to unborn babies. If a woman gets .htm rubella in the early months of her pregnancy, there is an 80% chance that www.cdc.gov/vaccines/vpd her baby will be born with birth defects. Babies may be born deaf or blind. They may have damaged hearts or small brains. Many are mentally -vac/rubella/default.htm retarded. Miscarriages are also common among women who get rubella while they are pregnant. What are the symptoms? The symptoms of Rubella include a slight fever that lasts for about 24 hours, and a rash on the face and neck that lasts 2 or 3 days. The rash is pink or light red spots that may merge to form splotches. -

The Effects of Atopy and Asthma on in Vivo Human Nasal Responses to Toll
The effects of atopy and asthma on in vivo human nasal responses to Toll-like receptor agonists Dr Akhilesh Jha A thesis submitted for the degree of Doctor of Philosophy (PhD) Centre for Respiratory Infection National Heart and Lung Institute (NHLI), St Mary’s Campus Imperial College London, Norfolk Place, London W2 1PG July 2018 Abstract Acute respiratory viral infections cause significant morbidity and mortality, especially in vulnerable individuals, and it is important to study viral pathogenesis and the host immune response in humans. Toll-like receptors (TLRs) play a critical role in the detection of viral nucleic acids, and airway TLR receptors respond to nucleic acid patterns in the RNA viruses that cause respiratory infections. However, a reliable method of measuring mucosal innate immune responses to viral infections is lacking. TLR3 agonists (poly(I:C) and poly-ICLC) and the combined TLR7/8 agonist (resiquimod, R848) are synthetic analogues of double stranded RNA (dsRNA) and single stranded RNA (ssRNA) respectively. Nasal challenge with these TLR agonists was carried out, and serial sampling using nasosorption and nasal curettage was performed. Mucosal immune responses were measured and the effect of different host factors (e.g. asthma) on these responses was studied. Poly(I:C) and poly-ICLC were well tolerated but failed to induce significant and reliable nasal mucosal innate immune responses. R848 at a higher dose (10 µg/100 µL per nostril) induced significant mucosal interferon and cytokine responses but caused mild to moderate flu-like symptoms in three out of nine volunteers. A lower dose of R848 (0.02 µg/kg/100 µL, mean dose 1.5 µg/100 µL) was subsequently utilised in three groups of volunteers: healthy non-atopic (n=12), allergic rhinitis (n=12) and allergic asthma (n=11). -
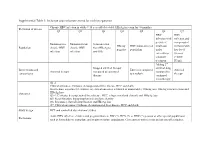
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

Bioinformatics Review of the Role of HSV-1 in Alzheimer's Disease
Advances in Alzheimer’s Disease, 2020, 9, 57-75 https://www.scirp.org/journal/aad ISSN Online: 2169-2467 ISSN Print: 2169-2459 Bioinformatics Review of the Role of HSV-1 in Alzheimer’s Disease Brian T. Reiss1, Meade C. Eggleston1, Arah C. Godbole1, Julia A. Marut1, Cecelia M. McCann1, Jessica A. Cottrell1, Tinchun Chu1, Sulie L. Chang1,2* 1Department of Biological Sciences, Seton Hall University, South Orange, NJ, USA 2Institute of Neuroimmune Pharmacology, Seton Hall University, South Orange, NJ, USA How to cite this paper: Reiss, B.T., Eg- Abstract gleston, M.C., Godbole, A.C., Marut, J.A., McCann, C.M., Cottrell, J.A., Chu, T.C. and Alzheimer’s disease (AD) is a neurodegenerative disease characterized by the Chang, S.L. (2020) Bioinformatics Review progressive loss of cognitive functions in affected individuals. Brain tissue of the Role of HSV-1 in Alzheimer’s Dis- pathology is associated with the formation of senile plaques which result from ease. Advances in Alzheimer’s Disease, 9, 57-75. the over-production of amyloid β (Aβ), due to the cleavage of a membrane https://doi.org/10.4236/aad.2020.93005 bound glycoprotein. It is unclear what causes AD and its associated patholo- gies, but age and genetic predisposition play an import role in the likelihood Received: June 11, 2020 Accepted: September 6, 2020 of disease development. Studies have shown that the reactivation of latent Published: September 9, 2020 herpes simplex virus 1 (HSV-1) infection can lead to the neuropathy of acute herpes simplex encephalitis (HSE), which causes similar symptoms to AD. -

Where Do We Stand After Decades of Studying Human Cytomegalovirus?
microorganisms Review Where do we Stand after Decades of Studying Human Cytomegalovirus? 1, 2, 1 1 Francesca Gugliesi y, Alessandra Coscia y, Gloria Griffante , Ganna Galitska , Selina Pasquero 1, Camilla Albano 1 and Matteo Biolatti 1,* 1 Laboratory of Pathogenesis of Viral Infections, Department of Public Health and Pediatric Sciences, University of Turin, 10126 Turin, Italy; [email protected] (F.G.); gloria.griff[email protected] (G.G.); [email protected] (G.G.); [email protected] (S.P.); [email protected] (C.A.) 2 Complex Structure Neonatology Unit, Department of Public Health and Pediatric Sciences, University of Turin, 10126 Turin, Italy; [email protected] * Correspondence: [email protected] These authors contributed equally to this work. y Received: 19 March 2020; Accepted: 5 May 2020; Published: 8 May 2020 Abstract: Human cytomegalovirus (HCMV), a linear double-stranded DNA betaherpesvirus belonging to the family of Herpesviridae, is characterized by widespread seroprevalence, ranging between 56% and 94%, strictly dependent on the socioeconomic background of the country being considered. Typically, HCMV causes asymptomatic infection in the immunocompetent population, while in immunocompromised individuals or when transmitted vertically from the mother to the fetus it leads to systemic disease with severe complications and high mortality rate. Following primary infection, HCMV establishes a state of latency primarily in myeloid cells, from which it can be reactivated by various inflammatory stimuli. Several studies have shown that HCMV, despite being a DNA virus, is highly prone to genetic variability that strongly influences its replication and dissemination rates as well as cellular tropism. In this scenario, the few currently available drugs for the treatment of HCMV infections are characterized by high toxicity, poor oral bioavailability, and emerging resistance. -

Activation of Anti-Hepatitis C Virus Responses Via Toll-Like Receptor 7
Activation of anti-hepatitis C virus responses via Toll-like receptor 7 Jongdae Lee*, Christina C. N. Wu*†, Ki Jeong Lee‡, Tsung-Hsien Chuang§, Kyoko Katakura*, Yu-Tsueng Liu*†, Michael Chan†, Rommel Tawatao†, Michelle Chung*, Carol Shen*, Howard B. Cottam†, Michael M. C. Lai‡, Eyal Raz*, and Dennis A. Carson†¶ *Department of Medicine, University of California at San Diego, La Jolla, CA 92093-0663; †Moores Cancer Center, University of California at San Diego Medical Center, La Jolla, CA 92093-0820; ‡Department of Molecular Microbiology and Immunology, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033; and §Department of Immunology, The Scripps Research Institute, La Jolla, CA 92037 Contributed by Dennis A. Carson, December 15, 2005 IFN-␣ is used to suppress the replication of hepatitis C virus (HCV) in HCV replicon cells. The IRFs can both foster type I IFN in chronically infected patients with partial success. Here we production (18) and enhance the antiviral activity of these present evidence showing that a ligand of Toll-like receptor 7 cytokines (19). Thus, pharmacologic agents that activate IRFs in (TLR7) can induce anti-HCV immunity not only by IFN induction, but hepatocytes may exert direct anti-HCV effects, as well as po- also through an IFN-independent mechanism. Human hepatocyte tentiate IFN action. line Huh-7 carrying an HCV replicon expressed TLR7, and activation Pharmacologic inhibitors of the enzyme inosine monophos- of the receptor induced several antiviral genes including IFN phate dehydrogenase (IMPDH), including ribavirin and mizor- regulatory factor-7. Inhibitors of the enzyme inosine monophos- ibine, have been reported to have activity against RNA viruses phate dehydrogenase augmented both IFN-dependent and -inde- (18–20). -

Annual Report 2016
Annexes to the annual report of the European Medicines Agency 2016 Annex 1 – Members of the Management Board ............................................................... 2 Annex 2 - Members of the Committee for Medicinal Products for Human Use ...................... 4 Annex 3 – Members of the Pharmacovigilance Risk Assessment Committee ........................ 6 Annex 4 – Members of the Committee for Medicinal Products for Veterinary Use ................. 8 Annex 5 – Members of the Committee on Orphan Medicinal Products .............................. 10 Annex 6 – Members of the Committee on Herbal Medicinal Products ................................ 12 Annex 7 – Committee for Advanced Therapies .............................................................. 14 Annex 8 – Members of the Paediatric Committee .......................................................... 16 Annex 9 – Working parties and working groups ............................................................ 18 Annex 10 – CHMP opinions: initial evaluations and extensions of therapeutic indication ..... 24 Annex 10a – Guidelines and concept papers adopted by CHMP in 2016 ............................ 25 Annex 11 – CVMP opinions in 2016 on medicinal products for veterinary use .................... 33 Annex 11a – 2016 CVMP opinions on extensions of indication for medicinal products for veterinary use .......................................................................................................... 39 Annex 11b – Guidelines and concept papers adopted by CVMP in 2016 ........................... -

Naive Alum | IMQ-Cpg
US010485861B2 United States Patent (10 ) Patent No.: US 10,485,861 B2 Stary et al. (45 ) Date of Patent : Nov. 26 , 2019 (54 ) NANOPARTICLE -BASED COMPOSITIONS Related U.S. Application Data ( 71) Applicants : President and Fellows of Harvard (60 ) Provisional application No. 61/ 783,439 , filed on Mar. College , Cambridge , MA (US ) : 14 , 2013 . Massachusetts Institute of (51 ) Int. CI. Technology , Cambridge , MA (US ); The A61K 39/118 (2006.01 ) Brigham and Women's Hospital, Inc., A61K 45/06 ( 2006.01) Boston , MA (US ) (Continued ) (52 ) U.S. CI. (72 ) Inventors : Georg Stary, Brookline, MA (US ) ; CPC A61K 39/118 (2013.01 ) ; A61K 31/4745 Aleksandar Filip Radovic -Moreno , ( 2013.01 ) ; A61K 31/711 ( 2013.01) ; State College , PA (US ) ; Pamela A. (Continued ) Basto , Cambridge, MA (US ) ; Michael ( 58 ) Field of Classification Search N. Starnbach , Needham , MA (US ) ; CPC A61K 9/10 ; A61K 35/74 ; A61K 39/39 ; Robert S. Langer , Newton , MA (US ) ; A61K 39/522 ; A61K 39/55555 ; Omid C. Farokhzad , Waban , MA (US ) ; Ulrich Von Andrian , Chestnut (Continued ) Hill , MA (US ) ( 56 ) References Cited (73 ) Assignees: President and Fellows of Harvard U.S. PATENT DOCUMENTS College, Cambridge , MA (US ) ; Massachusetts Institute of 3,960,757 A 6/1976 Morishita et al . Technology , Cambridge, MA (US ) ; The 4,638,045 A 11/1987 Kohn et al . Brigham and Women's Hospital, Inc. , (Continued ) Boston , MA (US ) FOREIGN PATENT DOCUMENTS ( * ) Notice : Subject to any disclaimer, the term of this WO WO1997028259 8/1997 patent is extended or adjusted under 35 WO WO1998016247 4/1998 U.S.C. 154 ( b ) by 0 days . -

Breast Cancer Treatment with Imiquimod: Applying an Old Lotion to a New Disease
Author Manuscript Published OnlineFirst on November 21, 2012; DOI: 10.1158/1078-0432.CCR-12-3138 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Breast cancer treatment with imiquimod: Applying an old lotion to a new disease Holbrook Kohrt, MD PhD Stanford University Cancer Institute Department of Medicine, Division of Oncology Stanford, CA 94305 Title: 79 characters Abstract: 49 words Text: 1200 words References: 12 references The author has no conflicts of interest. Running Title: Applying an old lotion to a new disease Downloaded from clincancerres.aacrjournals.org on September 25, 2021. © 2012 American Association for Cancer Research. Author Manuscript Published OnlineFirst on November 21, 2012; DOI: 10.1158/1078-0432.CCR-12-3138 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Abstract Over the prior two decades, imiquimod, a toll-like receptor 7 agonist, has been applied to nearly fifty clinical settings. Due to its immunomodulatory role, the topical cream today for the first time, is being applied to cutaneous breast cancer in pre-clinical models and in a Phase 2 clinical trial. Manuscript In this issue of Clinical Cancer Research, two sets of authors from Demaria’s group, Dewan et al. (1) and Adams et al. (2) detail in companion papers the anti-tumor efficacy of imiquimod in first, a preclinical breast cancer model in combination with radiation (1), and second, a Phase 2, breast cancer clinical trial assessing safety and immunologic activity (2). Fifteen years after first approval by the Food and Drug Administration (FDA), imiquimod remains the only approved, active toll-like receptor (TLR) agonist (3). -

Chronic Viral Infections Vs. Our Immune System: Revisiting Our View of Viruses As Pathogens
Chronic Viral Infections vs. Our Immune System: Revisiting our view of viruses as pathogens Tiffany A. Reese Assistant Professor Departments of Immunology and Microbiology Challenge your idea of classic viral infection and disease • Define the microbiome and the virome • Brief background on persistent viruses • Illustrate how viruses change disease susceptibility – mutualistic symbiosis – gene + virus = disease phenotype – virome in immune responses Bacteria-centric view of the microbiome The microbiome defined Definition of microbiome – Merriam-Webster 1 :a community of microorganisms (such as bacteria, fungi, and viruses) that inhabit a particular environment and especially the collection of microorganisms living in or on the human body 2 :the collective genomes of microorganisms inhabiting a particular environment and especially the human body Virome Ø Viral component of the microbiome Ø Includes both commensal and pathogenic viruses Ø Viruses that infect host cells Ø Virus-derived elements in host chromosomes Ø Viruses that infect other organisms in the body e.g. phage/bacteria Viruses are everywhere! • “intracellular parasites with nucleic acids that are capable of directing their own replication and are not cells” – Roossinck, Nature Reviews Microbiology 2011. • Viruses infect all living things. • We are constantly eating and breathing viruses from our environment • Only a small subset of viruses cause disease. • We even carry viral genomes as part of our own genetic material! Diverse viruses all over the body Adenoviridae Picornaviridae