Activation of Anti-Hepatitis C Virus Responses Via Toll-Like Receptor 7
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Newer Trends in the Management of Genital Herpes
Review NNewerewer trendstrends inin thethe mmanagementanagement ofof genitalgenital herpesherpes Article AAmiyamiya KKumarumar NNath,ath, DDevinderevinder MohanMohan ThappaThappa Department of Dermatology ABSTRACT and STD, Jawaharlal Institute of Postgraduate Medical Management of genital herpes is complex. Apart from using the standard antivirals, an Education and Research (JIPMER), Pondicherry - 605 ideal management protocol also needs to address various aspects of the disease, including 006, India the psychological morbidity. Oral acyclovir, valacyclovir or famciclovir are recommended for routine use. Long-term suppressive therapy is effective in reducing the number of AAddressddress forfor ccorrespondence:orrespondence: recurrences and the risk of transmission to others. Severe or disseminated disease may Dr. Devinder Mohan Thappa, require intravenous therapy. Resistant cases are managed with foscarnet or cidofovir. Genital Department of Dermatology herpes in human immunodeÞ ciency virus-infected individuals usually needs a longer duration and STD, JIPMER, of antiviral therapy along with continuation of highly active anti retroviral therapy (HAART). Pondicherry - 605 006, India. E-mail: [email protected] Genital herpes in late pregnancy increases the risk of neonatal herpes. Antiviral therapy and/or cesarean delivery are indicated depending on the clinical circumstance. Acyclovir appears to be safe in pregnancy. But, there is limited data regarding the use of valacyclovir and famciclovir in pregnancy. Neonatal herpes requires a higher dose of acyclovir given intravenously for a longer duration. Management of the sex partner, counseling and prevention advice are equally important in appropriate management of genital herpes. Vaccines till date have been marginally effective. Helicase–primase inhibitors, needle-free mucosal vaccine and a new microbicide product named VivaGel may become promising treatment options in the future. -

The Effects of Atopy and Asthma on in Vivo Human Nasal Responses to Toll
The effects of atopy and asthma on in vivo human nasal responses to Toll-like receptor agonists Dr Akhilesh Jha A thesis submitted for the degree of Doctor of Philosophy (PhD) Centre for Respiratory Infection National Heart and Lung Institute (NHLI), St Mary’s Campus Imperial College London, Norfolk Place, London W2 1PG July 2018 Abstract Acute respiratory viral infections cause significant morbidity and mortality, especially in vulnerable individuals, and it is important to study viral pathogenesis and the host immune response in humans. Toll-like receptors (TLRs) play a critical role in the detection of viral nucleic acids, and airway TLR receptors respond to nucleic acid patterns in the RNA viruses that cause respiratory infections. However, a reliable method of measuring mucosal innate immune responses to viral infections is lacking. TLR3 agonists (poly(I:C) and poly-ICLC) and the combined TLR7/8 agonist (resiquimod, R848) are synthetic analogues of double stranded RNA (dsRNA) and single stranded RNA (ssRNA) respectively. Nasal challenge with these TLR agonists was carried out, and serial sampling using nasosorption and nasal curettage was performed. Mucosal immune responses were measured and the effect of different host factors (e.g. asthma) on these responses was studied. Poly(I:C) and poly-ICLC were well tolerated but failed to induce significant and reliable nasal mucosal innate immune responses. R848 at a higher dose (10 µg/100 µL per nostril) induced significant mucosal interferon and cytokine responses but caused mild to moderate flu-like symptoms in three out of nine volunteers. A lower dose of R848 (0.02 µg/kg/100 µL, mean dose 1.5 µg/100 µL) was subsequently utilised in three groups of volunteers: healthy non-atopic (n=12), allergic rhinitis (n=12) and allergic asthma (n=11). -
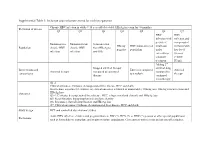
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

Bioinformatics Review of the Role of HSV-1 in Alzheimer's Disease
Advances in Alzheimer’s Disease, 2020, 9, 57-75 https://www.scirp.org/journal/aad ISSN Online: 2169-2467 ISSN Print: 2169-2459 Bioinformatics Review of the Role of HSV-1 in Alzheimer’s Disease Brian T. Reiss1, Meade C. Eggleston1, Arah C. Godbole1, Julia A. Marut1, Cecelia M. McCann1, Jessica A. Cottrell1, Tinchun Chu1, Sulie L. Chang1,2* 1Department of Biological Sciences, Seton Hall University, South Orange, NJ, USA 2Institute of Neuroimmune Pharmacology, Seton Hall University, South Orange, NJ, USA How to cite this paper: Reiss, B.T., Eg- Abstract gleston, M.C., Godbole, A.C., Marut, J.A., McCann, C.M., Cottrell, J.A., Chu, T.C. and Alzheimer’s disease (AD) is a neurodegenerative disease characterized by the Chang, S.L. (2020) Bioinformatics Review progressive loss of cognitive functions in affected individuals. Brain tissue of the Role of HSV-1 in Alzheimer’s Dis- pathology is associated with the formation of senile plaques which result from ease. Advances in Alzheimer’s Disease, 9, 57-75. the over-production of amyloid β (Aβ), due to the cleavage of a membrane https://doi.org/10.4236/aad.2020.93005 bound glycoprotein. It is unclear what causes AD and its associated patholo- gies, but age and genetic predisposition play an import role in the likelihood Received: June 11, 2020 Accepted: September 6, 2020 of disease development. Studies have shown that the reactivation of latent Published: September 9, 2020 herpes simplex virus 1 (HSV-1) infection can lead to the neuropathy of acute herpes simplex encephalitis (HSE), which causes similar symptoms to AD. -

Annual Report 2016
Annexes to the annual report of the European Medicines Agency 2016 Annex 1 – Members of the Management Board ............................................................... 2 Annex 2 - Members of the Committee for Medicinal Products for Human Use ...................... 4 Annex 3 – Members of the Pharmacovigilance Risk Assessment Committee ........................ 6 Annex 4 – Members of the Committee for Medicinal Products for Veterinary Use ................. 8 Annex 5 – Members of the Committee on Orphan Medicinal Products .............................. 10 Annex 6 – Members of the Committee on Herbal Medicinal Products ................................ 12 Annex 7 – Committee for Advanced Therapies .............................................................. 14 Annex 8 – Members of the Paediatric Committee .......................................................... 16 Annex 9 – Working parties and working groups ............................................................ 18 Annex 10 – CHMP opinions: initial evaluations and extensions of therapeutic indication ..... 24 Annex 10a – Guidelines and concept papers adopted by CHMP in 2016 ............................ 25 Annex 11 – CVMP opinions in 2016 on medicinal products for veterinary use .................... 33 Annex 11a – 2016 CVMP opinions on extensions of indication for medicinal products for veterinary use .......................................................................................................... 39 Annex 11b – Guidelines and concept papers adopted by CVMP in 2016 ........................... -

Naive Alum | IMQ-Cpg
US010485861B2 United States Patent (10 ) Patent No.: US 10,485,861 B2 Stary et al. (45 ) Date of Patent : Nov. 26 , 2019 (54 ) NANOPARTICLE -BASED COMPOSITIONS Related U.S. Application Data ( 71) Applicants : President and Fellows of Harvard (60 ) Provisional application No. 61/ 783,439 , filed on Mar. College , Cambridge , MA (US ) : 14 , 2013 . Massachusetts Institute of (51 ) Int. CI. Technology , Cambridge , MA (US ); The A61K 39/118 (2006.01 ) Brigham and Women's Hospital, Inc., A61K 45/06 ( 2006.01) Boston , MA (US ) (Continued ) (52 ) U.S. CI. (72 ) Inventors : Georg Stary, Brookline, MA (US ) ; CPC A61K 39/118 (2013.01 ) ; A61K 31/4745 Aleksandar Filip Radovic -Moreno , ( 2013.01 ) ; A61K 31/711 ( 2013.01) ; State College , PA (US ) ; Pamela A. (Continued ) Basto , Cambridge, MA (US ) ; Michael ( 58 ) Field of Classification Search N. Starnbach , Needham , MA (US ) ; CPC A61K 9/10 ; A61K 35/74 ; A61K 39/39 ; Robert S. Langer , Newton , MA (US ) ; A61K 39/522 ; A61K 39/55555 ; Omid C. Farokhzad , Waban , MA (US ) ; Ulrich Von Andrian , Chestnut (Continued ) Hill , MA (US ) ( 56 ) References Cited (73 ) Assignees: President and Fellows of Harvard U.S. PATENT DOCUMENTS College, Cambridge , MA (US ) ; Massachusetts Institute of 3,960,757 A 6/1976 Morishita et al . Technology , Cambridge, MA (US ) ; The 4,638,045 A 11/1987 Kohn et al . Brigham and Women's Hospital, Inc. , (Continued ) Boston , MA (US ) FOREIGN PATENT DOCUMENTS ( * ) Notice : Subject to any disclaimer, the term of this WO WO1997028259 8/1997 patent is extended or adjusted under 35 WO WO1998016247 4/1998 U.S.C. 154 ( b ) by 0 days . -

Breast Cancer Treatment with Imiquimod: Applying an Old Lotion to a New Disease
Author Manuscript Published OnlineFirst on November 21, 2012; DOI: 10.1158/1078-0432.CCR-12-3138 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Breast cancer treatment with imiquimod: Applying an old lotion to a new disease Holbrook Kohrt, MD PhD Stanford University Cancer Institute Department of Medicine, Division of Oncology Stanford, CA 94305 Title: 79 characters Abstract: 49 words Text: 1200 words References: 12 references The author has no conflicts of interest. Running Title: Applying an old lotion to a new disease Downloaded from clincancerres.aacrjournals.org on September 25, 2021. © 2012 American Association for Cancer Research. Author Manuscript Published OnlineFirst on November 21, 2012; DOI: 10.1158/1078-0432.CCR-12-3138 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Abstract Over the prior two decades, imiquimod, a toll-like receptor 7 agonist, has been applied to nearly fifty clinical settings. Due to its immunomodulatory role, the topical cream today for the first time, is being applied to cutaneous breast cancer in pre-clinical models and in a Phase 2 clinical trial. Manuscript In this issue of Clinical Cancer Research, two sets of authors from Demaria’s group, Dewan et al. (1) and Adams et al. (2) detail in companion papers the anti-tumor efficacy of imiquimod in first, a preclinical breast cancer model in combination with radiation (1), and second, a Phase 2, breast cancer clinical trial assessing safety and immunologic activity (2). Fifteen years after first approval by the Food and Drug Administration (FDA), imiquimod remains the only approved, active toll-like receptor (TLR) agonist (3). -

Time-Resolved Effect of Interferon-Alpha 2A on Activities of Nuclear Factor Kappa B, Pregnane X Receptor and on Drug Disposition Genes
pharmaceutics Article Time-Resolved Effect of Interferon-Alpha 2a on Activities of Nuclear Factor Kappa B, Pregnane X Receptor and on Drug Disposition Genes Dirk Theile , Lelia Wagner , Cindy Bay, Walter Emil Haefeli and Johanna Weiss * Department of Clinical Pharmacology and Pharmacoepidemiology, Heidelberg University Hospital, Im Neuenheimer Feld 410, 69120 Heidelberg, Germany; [email protected] (D.T.); [email protected] (L.W.); [email protected] (C.B.); [email protected] (W.E.H.) * Correspondence: [email protected]; Tel.: +49-6221-5639402 Abstract: Interferon-alpha (IFN-α) is suggested to cause pharmacokinetic drug interactions by lowering expression of drug disposition genes through affecting the activities of nuclear factor kappa B (NF-kB) and pregnane X receptor (PXR). The time-resolved impact of IFN-α 2a (1000 U/mL; 5000 U/mL; 2 h to 30 h) on the activities of NF-kB and PXR and mRNA expression (5000 U/mL; 24 h, 48 h) of selected drug disposition genes and on cytochrome P450 (CYP3A4) activity in LS180 cells (5000 U/mL; 24 h, 48 h) was evaluated using luciferase-based reporter gene assays, reverse transcription polymerase chain reaction, and luminescence-based CYP3A4 activity assays. The cross- talk between NF-kB activation and PXR suppression was evaluated by NF-kB blockage (10 µM parthenolide). IFN-α 2a initially (2 h, 6 h) enhanced NF-kB activity 2-fold and suppressed PXR Citation: Theile, D.; Wagner, L.; activity by 30%. mRNA of CYP3A4 was halved, whereas UGT1A1 was increased (1.35-fold) after Bay, C.; Haefeli, W.E.; Weiss, J. -

Immunosuppressant Ingredients, Immunostimulant Ingredients
Immunosuppressant Ingredients, Immunostimulant Ingredients Immunosuppressant Ingredients Immunosuppressant ingredients are chemical and medicinal agents used for the suppression and regulation of immune responses. Pharmacologically speaking, these agents have varied mecha- nisms of action, such as regulation of inflammatory gene expression, suppression of lymphocyte signal transduction, neutralization of cytokine activity, and inhibition lymphocyte proliferation by agent-induced cytotoxicity. Common agents include glucocorticoids, alkylating agents, metabolic antagonists, calcineurin inhibitors, T-cell suppressive agents, and cytokine inhibitors. 5-Aminosalicylic Acid 25g / 500g [A0317] 5-Aminosalicylic acid (5-ASA) is commonly used as a gastrointestinal anti-inflammatory agent. It has in vitro and in vivo pharmacologic effects that decrease leukotriene production, scavenge for free radicals, and inhibits leukocyte chemotaxis et al. Azathioprine 5g / 25g [A2069] Azathioprine is a prodrug of 6-mercaptopurine (6-MP), which inhibits the synthesis of purine ribonucleotides and DNA/RNA. Cyclophosphamide Monohydrate 5g / 25g [C2236] Cyclophosphamide is an antitumor alkylating reagent involved in the cross-linking of tumor cell DNA. Cyclosporin A 100mg / 1g [C2408] Cyclosporin A is a cyclic polypeptide immunosuppressant. It inhibits the activity of T-lymphocytes, and the phosphatase activity of calcineurin. Dimethyl Fumarate 25g / 500g [F0069] Dimethyl fumarate has neuroprotective and immunomodulating effects. Fingolimod Hydrochloride 200mg / 1g [F1018] Fingolimod (FTY720) is an immunomodulatory agent. It is an agonist at sphingosine 1-phosphate (S1P) receptors, and inhibits lymphocyte emigra- tion from lymphoid organs. Iguratimod 25mg / 250mg [I0945] Iguratimod (T-614) is an agent with anti-inflammatory and immunomodulatory activities. Its activity functions by the inhibiting production of immune globulin and inflammatory cytokines such as TNF-α, IL-1β and IL-6, and used as a disease modifying anti-rheumatic drug (DMARD). -

Rapid Host Immune Response and Viral Dynamics in Herpes Simplex Virus-2 Infection
PERSPECTIVE Rapid host immune response and viral dynamics in herpes simplex virus-2 infection Joshua T Schiffer1,2 & Lawrence Corey1–3 Herpes simplex virus-2 (HSV-2) is periodically shed throughout rate of HIV five- to tenfold14. Men and women who have acquired the human genital tract. Although a high viral load correlates HSV-2 have a two- to threefold increased risk of HIV-1 infection15,16. with the development of genital ulcers, shedding also In countries in which sexually active adults have a high prevalence commonly occurs even when ulcers are absent, allowing for of HSV-2 infection, or in subpopulations such as men who have silent transmission during coitus and contributing to high sex with men where HSV-2 infection is common, HSV-2 is a major seroprevalence of HSV-2 worldwide. Frequent viral reactivation epidemiological driver of HIV-1 epidemics16–18. occurs within ganglia despite diverse and complementary Here we describe how frequent reactivation and release of HSV-2 host and viral mechanisms that predispose toward latency, from latency within neurons, as well as highly dynamic interactions suggesting that viral replication may be constantly occurring between replicating HSV-2 and the host cell-mediated immune in a small minority of neurons at these sites. Within genital response in genital tissues, contribute to observed disease manifes- mucosa, the in vivo expansion and clearance rates of HSV-2 are tations, high global prevalence and enhanced HIV acquisition risk. extremely rapid. Resident dendritic cells and memory HSV-2 We also highlight the unique challenges that the kinetics of these specific T cells persist at prior sites of genital tract reactivation viral-host interactions pose for antiviral and vaccine development. -

Wo 2010/059253 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 27 May 2010 (27.05.2010) WO 2010/059253 A2 (51) International Patent Classification: 02140 (US). BERSHTEYN, Anna [US/US]; 25 Linden A61K 9/16 (2006.01) A61K 31/711 (2006.01) Avenue, Apartment 6, Somerville, MA 02143 (US). A61K 9/127 (2006.01) A61P 35/00 (2006.01) A61K 31/7105 (2006.01) (74) Agent: TREVISAN, Maria, A.; Wolf, Greenfield & Sacks, P.C , Federal Reserve Plaza, 600 Atlantic Avenue, (21) International Application Number: Boston, MA 02210-2206 (US). PCT/US2009/006290 (81) Designated States (unless otherwise indicated, for every (22) International Filing Date: kind of national protection available): AE, AG, AL, AM, 24 November 2009 (24.1 1.2009) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (25) Filing Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (26) Publication Language: English HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (30) Priority Data: ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, 61/200,1 60 24 November 2008 (24.1 1.2008) US NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (71) Applicant (for all designated States except US): MAS- SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, SACHUSETS INSTITUTE OF TECHNOLOGY [US/ TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

I Regulations
23.2.2007 EN Official Journal of the European Union L 56/1 I (Acts adopted under the EC Treaty/Euratom Treaty whose publication is obligatory) REGULATIONS COUNCIL REGULATION (EC) No 129/2007 of 12 February 2007 providing for duty-free treatment for specified pharmaceutical active ingredients bearing an ‘international non-proprietary name’ (INN) from the World Health Organisation and specified products used for the manufacture of finished pharmaceuticals and amending Annex I to Regulation (EEC) No 2658/87 THE COUNCIL OF THE EUROPEAN UNION, (4) In the course of three such reviews it was concluded that a certain number of additional INNs and intermediates used for production and manufacture of finished pharmaceu- ticals should be granted duty-free treatment, that certain of Having regard to the Treaty establishing the European Commu- these intermediates should be transferred to the list of INNs, nity, and in particular Article 133 thereof, and that the list of specified prefixes and suffixes for salts, esters or hydrates of INNs should be expanded. Having regard to the proposal from the Commission, (5) Council Regulation (EEC) No 2658/87 of 23 July 1987 on the tariff and statistical nomenclature and on the Common Customs Tariff (1) established the Combined Nomenclature Whereas: (CN) and set out the conventional duty rates of the Common Customs Tariff. (1) In the course of the Uruguay Round negotiations, the Community and a number of countries agreed that duty- (6) Regulation (EEC) No 2658/87 should therefore be amended free treatment should be granted to pharmaceutical accordingly, products falling within the Harmonised System (HS) Chapter 30 and HS headings 2936, 2937, 2939 and 2941 as well as to designated pharmaceutical active HAS ADOPTED THIS REGULATION: ingredients bearing an ‘international non-proprietary name’ (INN) from the World Health Organisation, specified salts, esters or hydrates of such INNs, and designated inter- Article 1 mediates used for the production and manufacture of finished products.