Multilocus Species Delimitation in a Complex of Morphologically Conserved Trapdoor Spiders (Mygalomorphae, Antrodiaetidae, Aliatypus)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biogeography of the Caribbean Cyrtognatha Spiders Klemen Čandek1,6,7, Ingi Agnarsson2,4, Greta J
www.nature.com/scientificreports OPEN Biogeography of the Caribbean Cyrtognatha spiders Klemen Čandek1,6,7, Ingi Agnarsson2,4, Greta J. Binford3 & Matjaž Kuntner 1,4,5,6 Island systems provide excellent arenas to test evolutionary hypotheses pertaining to gene fow and Received: 23 July 2018 diversifcation of dispersal-limited organisms. Here we focus on an orbweaver spider genus Cyrtognatha Accepted: 1 November 2018 (Tetragnathidae) from the Caribbean, with the aims to reconstruct its evolutionary history, examine Published: xx xx xxxx its biogeographic history in the archipelago, and to estimate the timing and route of Caribbean colonization. Specifcally, we test if Cyrtognatha biogeographic history is consistent with an ancient vicariant scenario (the GAARlandia landbridge hypothesis) or overwater dispersal. We reconstructed a species level phylogeny based on one mitochondrial (COI) and one nuclear (28S) marker. We then used this topology to constrain a time-calibrated mtDNA phylogeny, for subsequent biogeographical analyses in BioGeoBEARS of over 100 originally sampled Cyrtognatha individuals, using models with and without a founder event parameter. Our results suggest a radiation of Caribbean Cyrtognatha, containing 11 to 14 species that are exclusively single island endemics. Although biogeographic reconstructions cannot refute a vicariant origin of the Caribbean clade, possibly an artifact of sparse outgroup availability, they indicate timing of colonization that is much too recent for GAARlandia to have played a role. Instead, an overwater colonization to the Caribbean in mid-Miocene better explains the data. From Hispaniola, Cyrtognatha subsequently dispersed to, and diversifed on, the other islands of the Greater, and Lesser Antilles. Within the constraints of our island system and data, a model that omits the founder event parameter from biogeographic analysis is less suitable than the equivalent model with a founder event. -

Cryptic Species Delimitation in the Southern Appalachian Antrodiaetus
Cryptic species delimitation in the southern Appalachian Antrodiaetus unicolor (Araneae: Antrodiaetidae) species complex using a 3RAD approach Lacie Newton1, James Starrett1, Brent Hendrixson2, Shahan Derkarabetian3, and Jason Bond4 1University of California Davis 2Millsaps College 3Harvard University 4UC Davis May 5, 2020 Abstract Although species delimitation can be highly contentious, the development of reliable methods to accurately ascertain species boundaries is an imperative step in cataloguing and describing Earth's quickly disappearing biodiversity. Spider species delimi- tation remains largely based on morphological characters; however, many mygalomorph spider populations are morphologically indistinguishable from each other yet have considerable molecular divergence. The focus of our study, Antrodiaetus unicolor species complex which contains two sympatric species, exhibits this pattern of relative morphological stasis with considerable genetic divergence across its distribution. A past study using two molecular markers, COI and 28S, revealed that A. unicolor is paraphyletic with respect to A. microunicolor. To better investigate species boundaries in the complex, we implement the cohesion species concept and employ multiple lines of evidence for testing genetic exchangeability and ecological interchange- ability. Our integrative approach includes extensively sampling homologous loci across the genome using a RADseq approach (3RAD), assessing population structure across their geographic range using multiple genetic clustering analyses that include STRUCTURE, PCA, and a recently developed unsupervised machine learning approach (Variational Autoencoder). We eval- uate ecological similarity by using large-scale ecological data for niche-based distribution modeling. Based on our analyses, we conclude that this complex has at least one additional species as well as confirm species delimitations based on previous less comprehensive approaches. -

Final Project Completion Report
CEPF SMALL GRANT FINAL PROJECT COMPLETION REPORT Organization Legal Name: - Tarantula (Araneae: Theraphosidae) spider diversity, distribution and habitat-use: A study on Protected Area adequacy and Project Title: conservation planning at a landscape level in the Western Ghats of Uttara Kannada district, Karnataka Date of Report: 18 August 2011 Dr. Manju Siliwal Wildlife Information Liaison Development Society Report Author and Contact 9-A, Lal Bahadur Colony, Near Bharathi Colony Information Peelamedu Coimbatore 641004 Tamil Nadu, India CEPF Region: The Western Ghats Region (Sahyadri-Konkan and Malnad-Kodugu Corridors). 2. Strategic Direction: To improve the conservation of globally threatened species of the Western Ghats through systematic conservation planning and action. The present project aimed to improve the conservation status of two globally threatened (Molur et al. 2008b, Siliwal et al., 2008b) ground dwelling theraphosid species, Thrigmopoeus insignis and T. truculentus endemic to the Western Ghats through systematic conservation planning and action. Investment Priority 2.1 Monitor and assess the conservation status of globally threatened species with an emphasis on lesser-known organisms such as reptiles and fish. The present project was focused on an ignored or lesser-known group of spiders called Tarantulas/ Theraphosid spiders and provided valuable information on population status and potential conservation sites in Uttara Kannada district, which will help in future monitoring and assessment of conservation status of the two globally threatened theraphosid species T. insignis and Near Threatened T. truculentus. Investment Priority 2.3. Evaluate the existing protected area network for adequate globally threatened species representation and assess effectiveness of protected area types in biodiversity conservation. -

Tarantulas in the Pacific Northwest1
WSU Puyallup REC PLS-108 Updated July 2003 Tarantulas in the Pacific Northwest1 Tarantulas (Fig. 1) in the Pacific Northwest? Well, maybe not like the hairy monsters of the tropics, but some very interesting "atypical" species do occur here. Our species belong to the family Antrodiaetidae. One of our most common spiders is the folding-door spider, Antrodiaetus pacificus (Simon). It is a fairly large species, females ranging from 11 to 13 millimeters in length, males slightly smaller. They are generally dark brown to almost black in color with the abdomen purplish brown. Males are characterized by their long legs, slim bodies, and three tergites (hardened plates) on the abdomen. Females (Fig. 2) are more robust with only one tergite. These spiders excavate burrows in the soil or in damp, rotten wood, digging with a row of spines on each chelicer, known as a ratellum. The six to ten inch deep vertical shafts are lined with silk. The webbing extends beyond ground level as a short collar of camouflaged silk. The turret’s two sides may be drawn in by the occupant, forming two "doors" which meet in the middle. At night, Antrodiaetus assumes a foraging posture with its pedipalps and first pair of legs just touching the rim of silk at the mouth of the tube. In this position, the folding door spider can readily detect an insect moving above ground. The spider will leap out of its burrow with lightning speed, seize its victim, and drop back down, like a terrorizing Jack-in-the-box. When finished with its meal, it will add the insect's dry, dismembered body to a silk-covered trash pile at the bottom of its burrow. -
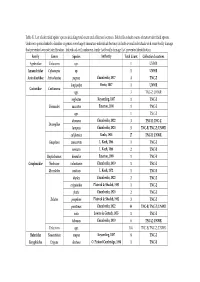
Table S1. List of Identified Spider Species Including Total Count and Collection Locations. Bold Cells Include Counts of Mature Identified Species
Table S1. List of identified spider species including total count and collection locations. Bold cells include counts of mature identified species. Unknown species linked to families or genera were largely immature individuals but may include several individuals with some bodily damage that prevented accurate identification. Individuals with unknown family had bodily damage that prevented identification. Family Genus Species Authority Total Count Collection Locations Agelenidae Unknown spp. 1 UNWR Amaurobiidae Cybaeopsis sp. 1 UNWR Antrodiaetidae Antrodiaetus pugnax Chamberlin, 1917 4 TNC-Z longipalpa Hentz, 1847 1 UNWR Corinnidae Castianeira spp. 3 TNC-Z; UNWR neglectus Keyserling, 1887 1 TNC-Z Drassodes saccatus Emerton, 1890 1 TNC-Z spp. 1 TNC-Z dromeus Chamberlin, 1922 3 TNC-B; TNC-Z Drassyllus lamprus Chamberlin, 1920 3 TNC-B; TNC-Z; UNWR californica Banks, 1904 17 TNC-B; UNWR Gnaphosa muscorum L. Koch, 1866 1 TNC-Z sericata L. Koch, 1866 2 TNC-B Haplodrassus hiemalis Emerton, 1909 1 TNC-B Gnaphosidae Nodocion voluntaries Chamberlin, 1919 1 TNC-Z Urozelotes rusticus L. Koch, 1872 1 TNC-B duplex Chamberlin, 1922 2 TNC-Z exiguioides Platnick & Shadab, 1983 1 TNC-Z fratis Chamberlin, 1920 2 TNC-Z Zelotes josephine Platnick & Shadab, 1983 3 TNC-Z puritanus Chamberlin, 1922 44 TNC-B; TNC-Z; UNWR sula Lowrie & Gertsch, 1955 1 TNC-Z tubuous Chamberlin, 1919 6 TNC-Z; UNWR Unknown spp. 184 TNC-B; TNC-Z; UNWR Hahniidae Neoantistea magna Keyserling, 1887 8 TNC-Z Linyphiidae Erigone dentosa O. Pickard-Cambridge, 1894 1 TNC-B spp. 1 TNC-Z Unknown spp. 13 TNC-Z; UNWR mccooki Montgomery, 1904 95 TNC-B; TNC-Z; UNWR Schizocosa minnesotensis Gertsch, 1934 25 TNC-B Lycosidae spp. -

Species Are Hypotheses: Avoid Connectivity Assessments Based on Pillars of Sand Eric Pante, Nicolas Puillandre, Amélia Viricel, Sophie Arnaud-Haond, D
Species are hypotheses: avoid connectivity assessments based on pillars of sand Eric Pante, Nicolas Puillandre, Amélia Viricel, Sophie Arnaud-Haond, D. Aurelle, Magalie Castelin, Anne Chenuil, Christophe Destombe, Didier Forcioli, Myriam Valero, et al. To cite this version: Eric Pante, Nicolas Puillandre, Amélia Viricel, Sophie Arnaud-Haond, D. Aurelle, et al.. Species are hypotheses: avoid connectivity assessments based on pillars of sand. Molecular Ecology, Wiley, 2015, 24 (3), pp.525-544. hal-02002440 HAL Id: hal-02002440 https://hal.archives-ouvertes.fr/hal-02002440 Submitted on 31 Jan 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Molecular Ecology Species are hypotheses : avoid basing connectivity assessments on pillars of sand. Journal:For Molecular Review Ecology Only Manuscript ID: Draft Manuscript Type: Invited Reviews and Syntheses Date Submitted by the Author: n/a Complete List of Authors: Pante, Eric; UMR 7266 CNRS - Université de La Rochelle, Puillandre, Nicolas; MNHN, Systematique & Evolution Viricel, Amélia; UMR 7266 CNRS - -

Phylogenomic Analysis and Revised Classification of Atypoid Mygalomorph Spiders (Araneae, Mygalomorphae), with Notes on Arachnid Ultraconserved Element Loci
Phylogenomic analysis and revised classification of atypoid mygalomorph spiders (Araneae, Mygalomorphae), with notes on arachnid ultraconserved element loci Marshal Hedin1, Shahan Derkarabetian1,2,3, Adan Alfaro1, Martín J. Ramírez4 and Jason E. Bond5 1 Department of Biology, San Diego State University, San Diego, CA, United States of America 2 Department of Biology, University of California, Riverside, Riverside, CA, United States of America 3 Department of Organismic and Evolutionary Biology, Museum of Comparative Zoology, Harvard University, Cambridge, MA, United States of America 4 Division of Arachnology, Museo Argentino de Ciencias Naturales ``Bernardino Rivadavia'', Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Buenos Aires, Argentina 5 Department of Entomology and Nematology, University of California, Davis, CA, United States of America ABSTRACT The atypoid mygalomorphs include spiders from three described families that build a diverse array of entrance web constructs, including funnel-and-sheet webs, purse webs, trapdoors, turrets and silken collars. Molecular phylogenetic analyses have generally supported the monophyly of Atypoidea, but prior studies have not sampled all relevant taxa. Here we generated a dataset of ultraconserved element loci for all described atypoid genera, including taxa (Mecicobothrium and Hexurella) key to understanding familial monophyly, divergence times, and patterns of entrance web evolution. We show that the conserved regions of the arachnid UCE probe set target exons, such that it should be possible to combine UCE and transcriptome datasets in arachnids. We also show that different UCE probes sometimes target the same protein, and under the matching parameters used here show that UCE alignments sometimes include non- Submitted 1 February 2019 orthologs. Using multiple curated phylogenomic matrices we recover a monophyletic Accepted 28 March 2019 Published 3 May 2019 Atypoidea, and reveal that the family Mecicobothriidae comprises four separate and divergent lineages. -

Folding Door Spider
Folding Door Spider Order: Arachnida Family: Antrodiaetidae Genus/species: Antrodiaetus species Description: The folding door spider is tan to chestnut brown about ¼” – 1 ½ “long. They have rakelike rows of spines on each jaw, or chelicera, which they use for digging. They have 3 claws on each foot. Folding door spiders have 1-4 small crescent shaped sclerites (hardened plates) on the top of the abdomen. They have 6 eyes in two groups of three. Habitat: Folding door spiders excavate burrows in the ground that they line with silk. At the entrance they construct a collapsible “trap door”. When active they open their trapdoor at night and wait just inside the entrance to strike at passing prey. The spider detects the prey by sensing vibrations. When mature, males abandon their burrows during rainy periods and wander in search of females. Life Cycle: Most spiders live 1 to 2 years. The adult male begins courtship after his palp is filled with sperm and he has found a female. Soon after mating the male dies. After a week or more the mated female deposits her eggs in a silken sac. Weeks later, or sometimes not until the following spring, the young spiderlings emerge. Economic Importance & Management: All spiders are voracious predators. Folding Door spiders feed on insects, and are considered highly beneficial because they keep the burgeoning insect populations in check. References: The Audubon Society Field Guide to NA Lorus & Marjery Milne Spiders! Usually helpful, sometimes a nuisance, Jean Natter Spiders of NA-identification manual, D. Ubick, P. Paquin P.E .Cushing and V. -

Epigeic Spider (Araneae) Diversity and Habitat Distributions in Kings
Clemson University TigerPrints All Theses Theses 5-2011 Epigeic Spider (Araneae) Diversity and Habitat Distributions in Kings Mountain National Military Park, South Carolina Sarah Stellwagen Clemson University, [email protected] Follow this and additional works at: https://tigerprints.clemson.edu/all_theses Part of the Entomology Commons Recommended Citation Stellwagen, Sarah, "Epigeic Spider (Araneae) Diversity and Habitat Distributions in Kings Mountain National Military Park, South Carolina" (2011). All Theses. 1091. https://tigerprints.clemson.edu/all_theses/1091 This Thesis is brought to you for free and open access by the Theses at TigerPrints. It has been accepted for inclusion in All Theses by an authorized administrator of TigerPrints. For more information, please contact [email protected]. EPIGEIC SPIDER (ARANEAE) DIVERSITY AND HABITAT DISTRIBUTIONS IN KINGS MOUNTAIN NATIONAL MILITARY PARK, SOUTH CAROLINA ______________________________ A Thesis Presented to the Graduate School of Clemson University _______________________________ In Partial Fulfillment of the Requirements for the Degree Masters of Science Entomology _______________________________ by Sarah D. Stellwagen May 2011 _______________________________ Accepted by: Dr. Joseph D. Culin, Committee Chair Dr. Eric Benson Dr. William Bridges ABSTRACT This study examined the epigeic spider fauna in Kings Mountain National Military Park. The aim of this study is to make this information available to park management for use in the preservation of natural resources. Pitfall trapping was conducted monthly for one year in three distinct habitats: riparian, forest, and ridge-top. The study was conducted from August 2009 to July 2010. One hundred twenty samples were collected in each site. Overall, 289 adult spiders comprising 66 species were collected in the riparian habitat, 345 adult comprising 57 species were found in the forest habitat, and 240 adults comprising 47 species were found in the ridge-top habitat. -

Cladistic Analysis of the Atypoides Plus Antrodiaetus Lineage of Mygalomorph Spiders (Araneae, Antrodiaetidae )
1996 . The Journal of Arachnology 24 :201–21 3 CLADISTIC ANALYSIS OF THE ATYPOIDES PLUS ANTRODIAETUS LINEAGE OF MYGALOMORPH SPIDERS (ARANEAE, ANTRODIAETIDAE ) Jeremy A . Miller and Frederick A . Coyle : Department of Biology, Western Carolina University, Cullowhee, North Carolina 28723 USA ABSTRACT . Cladistic analyses of the antrodiaetid spider genera Atypoides O .P.-Cambridge 1883 and Antrodiaetus Ausserer 1871 yield a much more completely resolved phylogeny than that proposed by Coyle in 1971 . Twenty-nine potentially informative characters were used in the analyses, which wer e performed using PAUPs a posteriori weighting options . Three independent analyses were performed , each with a different outgroup . These outgroups were 1) the antrodiaetid genus Aliatypus Smith 1908, the putative sister group of Atypoides plus Antrodiaetus, 2) Aliatypus gulosus Coyle 1974, the most primitive Aliatypus species, and 3) a hypothetical ancestral taxon based on character states found in Aliatypus an d the Atypidae, the latter being the putative sister group of the antrodiaetids . These three analyses produced a total of eight most parsimonious trees which support the following principal conclusions : 1) Atypoides, as defined by Coyle, is paraphyletic (Atypoides riversi O . P.-Cambridge 1883 plus At. gertschi Coyle 1968 share with Antrodiaetus a common ancestor not shared with At . hadros Coyle 1968). 2) Antrodiaetus roretzi (L. Koch 1878) is a relict species which shares a unique common ancestor with all other Antro- diaetus species . 3) Coyles unicolor group of nine Antrodiaetus species is paraphyletic ; six of these form a recently-derived Glade, (Antrodiaetus occultus Coyle 1971 (An . yesoensis [Uyemura 1942], An. cerberu s Coyle 1971, (An. montanus [Chamberlin Ivie 1933], (An. -

A Reconsideration of the Classification of the Spider Infraorder Mygalomorphae (Arachnida: Araneae) Based on Three Nuclear Genes and Morphology
A Reconsideration of the Classification of the Spider Infraorder Mygalomorphae (Arachnida: Araneae) Based on Three Nuclear Genes and Morphology Jason E. Bond1*, Brent E. Hendrixson2, Chris A. Hamilton1, Marshal Hedin3 1 Department of Biological Sciences and Auburn University Museum of Natural History, Auburn University, Auburn, Alabama, United States of America, 2 Department of Biology, Millsaps College, Jackson, Mississippi, United States of America, 3 Department of Biology, San Diego State University, San Diego, California, United States of America Abstract Background: The infraorder Mygalomorphae (i.e., trapdoor spiders, tarantulas, funnel web spiders, etc.) is one of three main lineages of spiders. Comprising 15 families, 325 genera, and over 2,600 species, the group is a diverse assemblage that has retained a number of features considered primitive for spiders. Despite an evolutionary history dating back to the lower Triassic, the group has received comparatively little attention with respect to its phylogeny and higher classification. The few phylogenies published all share the common thread that a stable classification scheme for the group remains unresolved. Methods and Findings: We report here a reevaluation of mygalomorph phylogeny using the rRNA genes 18S and 28S, the nuclear protein-coding gene EF-1c, and a morphological character matrix. Taxon sampling includes members of all 15 families representing 58 genera. The following results are supported in our phylogenetic analyses of the data: (1) the Atypoidea (i.e., antrodiaetids, atypids, and mecicobothriids) is a monophyletic group sister to all other mygalomorphs; and (2) the families Mecicobothriidae, Hexathelidae, Cyrtaucheniidae, Nemesiidae, Ctenizidae, and Dipluridae are not monophyletic. The Microstigmatidae is likely to be subsumed into Nemesiidae. -

Annual Report 2011–2012
Annual Report 2011–2012 San Diego Natural History Museum Our Mission To interpret the natural world through research, education and exhibits; to promote understanding of the evolution and diversity of southern California and the peninsula of Baja California; and to inspire in all a respect for nature and the environment. October 2012 Dear Museum Friends, We close the 2011–12 fi scal year with renewed optimism and a collective sigh of relief. Over this past year we continued to realize the benefi ts of the many diffi cult decisions made in years 2008 through 2010. The year 2008 felt like the perfect economic storm, but critical analysis over the period of the recession and nimble strategic management last fall ushered in a new day when the Museum secured Titanic: The Artifact Exhibition which generated a critical revenue stream. Coupled with our self-imposed austerity measures, this year’s strong admissions and attendance numbers have helped to bring the Museum back to a fi rm fi nancial footing. This begs an important question: why is the San Diego Natural History Museum doing an exhibition on the Titanic? What does Titanic have to do with natural history? Our strategy for traveling exhibitions stems from a philosophy fi rst espoused by the Legler Benbough Foundation. When the Benbough Foundation funded the traveling exhibition hall that bears their name, the Foundation did so with the goal of “providing opportunities for San Diegans that would be otherwise unavailable.” As an important member of the San Diego regional arts and culture community, we are providing opportunities to experience the fi nest exhibitions available regionally, nationally, and internationally.