Understanding the Histone DNA Repair Code: H4k20me2 Makes Its Mark
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Predicting Double-Strand DNA Breaks Using Epigenome Marks Or DNA at Kilobase Resolution
bioRxiv preprint doi: https://doi.org/10.1101/149039; this version posted June 12, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Predicting double-strand DNA breaks using epigenome marks or DNA at kilobase resolution Rapha¨elMourad1 and Olivier Cuvier1 April 10, 2017 1 Laboratoire de Biologie Mol´eculaireEucaryote (LBME), CNRS, Universit´ePaul Sabatier (UPS), 31000 Toulouse, France Abstract Double-strand breaks (DSBs) result from the attack of both DNA strands by multiple sources, includ- ing exposure to ionizing radiation or reactive oxygen species. DSBs can cause abnormal chromosomal rearrangements which are linked to cancer development, and hence represent an important issue. Recent techniques allow the genome-wide mapping of DSBs at high resolution, enabling the comprehensive study of DSB origin. However these techniques are costly and challenging. Hence we devised a computational approach to predict DSBs using the epigenomic and chromatin context, for which public data are available from the ENCODE project. We achieved excellent prediction accuracy (AUC = 0:97) at high resolution (< 1 kb), and showed that only chromatin accessibility and H3K4me1 mark were sufficient for highly accurate prediction (AUC = 0:95). We also demonstrated the better sensitivity of DSB predictions com- pared to BLESS experiments. We identified chromatin accessibility, activity and long-range contacts as best predictors. In addition, our work represents the first step toward unveiling the "cis-DNA repairing" code underlying DSBs, paving the way for future studies of cis-elements involved in DNA damage and repair. -

Application of Histone Modification-Specific Interaction Domains As an Alternative to Antibodies
Downloaded from genome.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press Method Application of histone modification-specific interaction domains as an alternative to antibodies Goran Kungulovski,1 Ina Kycia,1,4 Raluca Tamas,1 Renata Z. Jurkowska,1 Srikanth Kudithipudi,1 Chisato Henry,2 Richard Reinhardt,3 Paul Labhart,2 and Albert Jeltsch1 1Institute of Biochemistry, Stuttgart University, 70569 Stuttgart, Germany; 2Active Motif, Carlsbad, California 92008, USA; 3Max-Planck-Genomzentrum Koln,€ 50829 Koln,€ Germany Post-translational modifications (PTMs) of histones constitute a major chromatin indexing mechanism, and their proper characterization is of highest biological importance. So far, PTM-specific antibodies have been the standard reagent for studying histone PTMs despite caveats such as lot-to-lot variability of specificity and binding affinity. Herein, we suc- cessfully employed naturally occurring and engineered histone modification interacting domains for detection and identification of histone PTMs and ChIP-like enrichment of different types of chromatin. Our results demonstrate that histone interacting domains are robust and highly specific reagents that can replace or complement histone modification antibodies. These domains can be produced recombinantly in Escherichia coli at low cost and constant quality. Protein design of reading domains allows for generation of novel specificities, addition of affinity tags, and preparation of PTM binding pocket variants as matching negative controls, which is not possible with antibodies. [Supplemental material is available for this article.] The unstructured N-terminal tails of histones protrude from the yielding false negative results. When undocumented, the cross- core nucleosome and harbor complex patterns of post-translational reactivity with related or unrelated marks and the combinatorial modifications (PTMs) (Kouzarides 2007; Margueron and Reinberg effect of neighboring marks compromise the application of anti- 2010; Bannister and Kouzarides 2011; Tan et al. -

Targeting the Methyltransferase SETD8 Impairs Tumor Cell Survival and Overcomes Drug Resistance Independently of P53 Status in Multiple Myeloma
bioRxiv preprint doi: https://doi.org/10.1101/776930; this version posted September 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Targeting the methyltransferase SETD8 impairs tumor cell survival and overcomes drug resistance independently of p53 status in multiple myeloma Laurie Herviou (1,2,4), Fanny Izard (3,4), Ouissem Karmous-Gadacha (2) , Claire Gourzones (1), Celine Bellanger (1), Eva Desmedt (5), Anqi Ma (6), Laure Vincent (7) , Guillaume Cartron (4,7), Karin Vanderkerken (5), Jian Jin (6), Elke De Bruyne (5), Charlotte Grimaud (3,4,8), Eric Julien (3,4,8 +) and Jérôme Moreaux (1,2,4+) (1) IGH, CNRS, Univ Montpellier, France (2) CHU Montpellier, Laboratory for Monitoring Innovative Therapies, Department of Biological Hematology, Montpellier, France (3) Institut de Recherche en Cancérologie de Montpellier (IRCM), INSERM U1194, Institut Régional du Cancer (ICM), Montpellier F-34298, France (4) University of Montpellier, Montpellier, F-34090, France (5) Department of Hematology and Immunology-Myeloma Center Brussels, Vrije Universiteit Brussel, Brussels, Belgium (6) Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, NeW York, NeW York 10029, United States. (7) CHU Montpellier, Department of Clinical Hematology, Montpellier, France (8) Centre National de la Recherche Scientifique (CNRS), F-34293, Montpellier, France + : co-last and corresponding authors ; corresponding authors: Eric Julien ([email protected]) and jérôme Moreaux ([email protected]). 1 bioRxiv preprint doi: https://doi.org/10.1101/776930; this version posted September 20, 2019. -

TRCP Promotes Cell Growth by Targeting PR-Set7/Set8 for Degradation
SCFβ-TRCP promotes cell growth by targeting PR-Set7/Set8 for degradation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Wang, Z., X. Dai, J. Zhong, H. Inuzuka, L. Wan, X. Li, L. Wang, et al. 2015. “SCFβ-TRCP promotes cell growth by targeting PR-Set7/Set8 for degradation.” Nature Communications 6 (1): 10185. doi:10.1038/ ncomms10185. http://dx.doi.org/10.1038/ncomms10185. Published Version doi:10.1038/ncomms10185 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:23993465 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ARTICLE Received 4 May 2015 | Accepted 12 Nov 2015 | Published 15 Dec 2015 DOI: 10.1038/ncomms10185 OPEN SCFb-TRCP promotes cell growth by targeting PR-Set7/Set8 for degradation Zhiwei Wang1,2,*, Xiangpeng Dai2,*, Jiateng Zhong2,3,*, Hiroyuki Inuzuka2, Lixin Wan2, Xiaoning Li2,4, Lixia Wang1, Xiantao Ye1, Liankun Sun4, Daming Gao2,5,LeeZou6 & Wenyi Wei2 The Set8/PR-Set7/KMT5a methyltransferase plays critical roles in governing transcriptional regulation, cell cycle progression and tumorigenesis. Although CRL4Cdt2 was reported to regulate Set8 stability, deleting the PIP motif only led to partial resistance to ultraviolet- induced degradation of Set8, indicating the existence of additional E3 ligase(s) controlling Set8 stability. Furthermore, it remains largely undefined how DNA damage-induced kinase cascades trigger the timely destruction of Set8 to govern tumorigenesis. -
H4K20 Monomethylation Faces the WNT
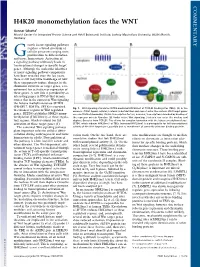
COMMENTARY H4K20 monomethylation faces the WNT Gunnar Schotta1 Munich Center for Integrated Protein Science and Adolf Butenandt Institute, Ludwig Maximilians University, 80336 Munich, Germany rowth factor signaling pathways regulate a broad spectrum of G cellular processes ranging from proliferation to differentiation and tissue homeostasis. Activation of a signaling pathway ultimately leads to transcriptional changes in specific target genes. Although the molecular identities of many signaling pathway components have been revealed over the last years, there is still very little knowledge of how these components induce changes in the chromatin structure of target genes, a re- quirement for activation or repression of these genes. A new link is provided by an interesting paper in PNAS that demon- strates that in the context of Wnt signaling the histone methyltransferase SETD8 (PR-SET7, KMT5a, SET8) is recruited Fig. 1. Wnt signaling stimulates SETD8-mediated H4K20me1 at TCF/LEF binding sites (TBEs). (A)Inthe to enhancer regions of Wnt-regulated absence of Wnt ligand, cellular β-catenin is destabilized and cannot enter the nucleus. Wnt target genes genes. SETD8 establishes H4K20 mono- are constitutively bound by TCF/LEF transcription factors; however, transcription is blocked by binding of methylation (H4K20me1) at these regula- the repressor protein Groucho. (B) Under active Wnt signaling, β-catenin can enter the nucleus and tory regions, which is crucial for full displace Groucho from TCF/LEF. This allows for complex formation with the histone methyltransferase activation of these target genes (1). SETD8, which induces H4K20me1 at TBEs. Increased H4K20me1 is a prerequisite for full transcriptional The canonical Wnt signaling pathway activity of the Wnt target gene, possibly due to recruitment of currently unknown binding proteins. -

Histone H4 Lysine 20 Mono-Methylation Directly Facilitates Chromatin Openness and Promotes Transcription of Housekeeping Genes
ARTICLE https://doi.org/10.1038/s41467-021-25051-2 OPEN Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes Muhammad Shoaib 1,8,9, Qinming Chen2,9, Xiangyan Shi 3, Nidhi Nair1, Chinmayi Prasanna 2, Renliang Yang2,4, David Walter1, Klaus S. Frederiksen 5, Hjorleifur Einarsson1, J. Peter Svensson 6, ✉ ✉ ✉ Chuan Fa Liu 2, Karl Ekwall6, Mads Lerdrup 7 , Lars Nordenskiöld 2 & Claus S. Sørensen 1 1234567890():,; Histone lysine methylations have primarily been linked to selective recruitment of reader or effector proteins that subsequently modify chromatin regions and mediate genome functions. Here, we describe a divergent role for histone H4 lysine 20 mono-methylation (H4K20me1) and demonstrate that it directly facilitates chromatin openness and accessibility by disrupting chromatin folding. Thus, accumulation of H4K20me1 demarcates highly accessible chromatin at genes, and this is maintained throughout the cell cycle. In vitro, H4K20me1-containing nucleosomal arrays with nucleosome repeat lengths (NRL) of 187 and 197 are less compact than unmethylated (H4K20me0) or trimethylated (H4K20me3) arrays. Concordantly, and in contrast to trimethylated and unmethylated tails, solid-state NMR data shows that H4K20 mono-methylation changes the H4 conformational state and leads to more dynamic histone H4-tails. Notably, the increased chromatin accessibility mediated by H4K20me1 facilitates gene expression, particularly of housekeeping genes. Altogether, we show how the methy- lation state of a single histone H4 residue operates as a focal point in chromatin structure control. While H4K20me1 directly promotes chromatin openness at highly transcribed genes, it also serves as a stepping-stone for H4K20me3-dependent chromatin compaction. -

Mechanism of CRL4 , a PCNA-Dependent E3 Ubiquitin Ligase
Downloaded from genesdev.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Mechanism of CRL4Cdt2, a PCNA-dependent E3 ubiquitin ligase Courtney G. Havens and Johannes C. Walter1 Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, USA Eukaryotic cell cycle transitions are driven by E3 ubiq- the ubiquitin is transferred from E1 to the cysteine of an uitin ligases that catalyze the ubiquitylation and destruc- ‘‘E2’’ ubiquitin-conjugating enzyme. Finally, the E2 in- tion of specific protein targets. For example, the anaphase- teracts with an ‘‘E3’’ ubiquitin ligase that also binds the promoting complex/cyclosome (APC/C) promotes the substrate. The juxtaposition of the substrate and the charged exit from mitosis via destruction of securin and mitotic E2 enzyme leads to ubiquitin transfer to the substrate. cyclins, whereas CRL1Skp2 allows entry into S phase by The specificity of ubiquitylation is encoded at the level of targeting the destruction of the cyclin-dependent kinase substrate recognition by the E3 enzymes (Ravid and (CDK) inhibitor p27. Recently, an E3 ubiquitin ligase Hochstrasser 2008); however, recently it has become called CRL4Cdt2 has been characterized, which couples clear that E2s can also contribute to processivity and proteolysis to DNA synthesis via an unusual mechanism specificity for ubiquitin chain nucleation and elongation that involves display of substrate degrons on the DNA (Jin et al. 2008; Ye and Rape 2009; Rodrigo-Brenni et al. polymerase processivity factor PCNA. Through its de- 2010; Saha et al. 2011; Wickliffe et al. 2011). Generally, struction of Cdt1, p21, and Set8, CRL4Cdt2 has emerged as the E3–substrate interaction involves the binding of a master regulator that prevents rereplication in S phase. -

Novel Pharmacological Maps of Protein Lysine Methyltransferases: Key for Target Deorphanization Obdulia Rabal* , Andrea Castellar and Julen Oyarzabal*
Rabal et al. J Cheminform (2018) 10:32 https://doi.org/10.1186/s13321-018-0288-5 RESEARCH ARTICLE Open Access Novel pharmacological maps of protein lysine methyltransferases: key for target deorphanization Obdulia Rabal* , Andrea Castellar and Julen Oyarzabal* Abstract Epigenetic therapies are being investigated for the treatment of cancer, cognitive disorders, metabolic alterations and autoinmune diseases. Among the diferent epigenetic target families, protein lysine methyltransferases (PKMTs), are especially interesting because it is believed that their inhibition may be highly specifc at the functional level. Despite its relevance, there are currently known inhibitors against only 10 out of the 50 SET-domain containing members of the PKMT family. Accordingly, the identifcation of chemical probes for the validation of the therapeutic impact of epigenetic modulation is key. Moreover, little is known about the mechanisms that dictate their substrate specifc- ity and ligand selectivity. Consequently, it is desirable to explore novel methods to characterize the pharmacologi- cal similarity of PKMTs, going beyond classical phylogenetic relationships. Such characterization would enable the prediction of ligand of-target efects caused by lack of ligand selectivity and the repurposing of known compounds against alternative targets. This is particularly relevant in the case of orphan targets with unreported inhibitors. Here, we frst perform a systematic study of binding modes of cofactor and substrate bound ligands with all available SET domain-containing PKMTs. Protein ligand interaction fngerprints were applied to identify conserved hot spots and contact-specifc residues across subfamilies at each binding site; a relevant analysis for guiding the design of novel, selective compounds. Then, a recently described methodology (GPCR-CoINPocket) that incorporates ligand contact information into classical alignment-based comparisons was applied to the entire family of 50 SET-containing proteins to devise pharmacological similarities between them. -

Histone 4 Lysine 20 Methylation: a Case for Neurodevelopmental Disease
biology Review Histone 4 Lysine 20 Methylation: A Case for Neurodevelopmental Disease Rochelle N. Wickramasekara and Holly A. F. Stessman * Department of Pharmacology, School of Medicine, Creighton University, Omaha, NE 68178, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-402-280-2255 Received: 28 December 2018; Accepted: 26 February 2019; Published: 3 March 2019 Abstract: Neurogenesis is an elegantly coordinated developmental process that must maintain a careful balance of proliferation and differentiation programs to be compatible with life. Due to the fine-tuning required for these processes, epigenetic mechanisms (e.g., DNA methylation and histone modifications) are employed, in addition to changes in mRNA transcription, to regulate gene expression. The purpose of this review is to highlight what we currently know about histone 4 lysine 20 (H4K20) methylation and its role in the developing brain. Utilizing publicly-available RNA-Sequencing data and published literature, we highlight the versatility of H4K20 methyl modifications in mediating diverse cellular events from gene silencing/chromatin compaction to DNA double-stranded break repair. From large-scale human DNA sequencing studies, we further propose that the lysine methyltransferase gene, KMT5B (OMIM: 610881), may fit into a category of epigenetic modifier genes that are critical for typical neurodevelopment, such as EHMT1 and ARID1B, which are associated with Kleefstra syndrome (OMIM: 610253) and Coffin-Siris syndrome (OMIM: 135900), respectively. Based on our current knowledge of the H4K20 methyl modification, we discuss emerging themes and interesting questions on how this histone modification, and particularly KMT5B expression, might impact neurodevelopment along with current challenges and potential avenues for future research. -

The Emerging Role for Cullin 4 Family of E3 Ligases in Tumorigenesis T ⁎ ⁎ Ji Chenga,B,1, Jianping Guob,1, Brian J
BBA - Reviews on Cancer 1871 (2019) 138–159 Contents lists available at ScienceDirect BBA - Reviews on Cancer journal homepage: www.elsevier.com/locate/bbacan Review The emerging role for Cullin 4 family of E3 ligases in tumorigenesis T ⁎ ⁎ Ji Chenga,b,1, Jianping Guob,1, Brian J. Northb, Kaixiong Taoa, Pengbo Zhouc, , Wenyi Weib, a Department of Gastrointestinal Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China b Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02215, USA c Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, 1300 York Ave., New York, NY 10065, USA ARTICLE INFO ABSTRACT Keywords: As a member of the Cullin-RING ligase family, Cullin-RING ligase 4 (CRL4) has drawn much attention due to its CRL4, Cullin 4 broad regulatory roles under physiological and pathological conditions, especially in neoplastic events. Based on E3 ligases evidence from knockout and transgenic mouse models, human clinical data, and biochemical interactions, we PROTACs summarize the distinct roles of the CRL4 E3 ligase complexes in tumorigenesis, which appears to be tissue- and Tumorigenesis context-dependent. Notably, targeting CRL4 has recently emerged as a noval anti-cancer strategy, including Targeted therapy thalidomide and its derivatives that bind to the substrate recognition receptor cereblon (CRBN), and anticancer sulfonamides that target DCAF15 to suppress the neoplastic proliferation of multiple myeloma and colorectal cancers, respectively. To this end, PROTACs have been developed as a group of engineered bi-functional che- mical glues that induce the ubiquitination-mediated degradation of substrates via recruiting E3 ligases, such as CRL4 (CRBN) and CRL2 (pVHL). -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase.